Suggestions or feedback?

MIT News | Massachusetts Institute of Technology
- Machine learning
- Social justice
- Black holes
- Classes and programs
Departments
- Aeronautics and Astronautics
- Brain and Cognitive Sciences
- Architecture
- Political Science
- Mechanical Engineering
Centers, Labs, & Programs
- Abdul Latif Jameel Poverty Action Lab (J-PAL)
- Picower Institute for Learning and Memory
- Lincoln Laboratory
- School of Architecture + Planning
- School of Engineering
- School of Humanities, Arts, and Social Sciences
- Sloan School of Management
- School of Science
- MIT Schwarzman College of Computing
Scientists discover how mutations in a language gene produce speech deficits
Press contact :, media download.

*Terms of Use:
Images for download on the MIT News office website are made available to non-commercial entities, press and the general public under a Creative Commons Attribution Non-Commercial No Derivatives license . You may not alter the images provided, other than to crop them to size. A credit line must be used when reproducing images; if one is not provided below, credit the images to "MIT."

Previous image Next image
Mutations of a gene called Foxp2 have been linked to a type of speech disorder called apraxia that makes it difficult to produce sequences of sound. A new study from MIT and National Yang Ming Chiao Tung University sheds light on how this gene controls the ability to produce speech.
In a study of mice, the researchers found that mutations in Foxp2 disrupt the formation of dendrites and neuronal synapses in the brain’s striatum, which plays important roles in the control of movement. Mice with these mutations also showed impairments in their ability to produce the high-frequency sounds that they use to communicate with other mice.
Those malfunctions arise because Foxp2 mutations prevent the proper assembly of motor proteins, which move molecules within cells, the researchers found.
“These mice have abnormal vocalizations, and in the striatum there are many cellular abnormalities,” says Ann Graybiel, an MIT Institute Professor, a member of MIT’s McGovern Institute for Brain Research, and an author of the paper . “This was an exciting finding. Who would have thought that a speech problem might come from little motors inside cells?”
Fu-Chin Liu PhD ’91, a professor at National Yang Ming Chiao Tung University in Taiwan, is the senior author of the study, which appears today in the journal Brain . Liu and Graybiel also worked together on a 2016 study of the potential link between Foxp2 and autism spectrum disorder. The lead authors of the new Brain paper are Hsiao-Ying Kuo and Shih-Yun Chen of National Yang Ming Chiao Tung University.
Speech control
Children with Foxp2-associated apraxia tend to begin speaking later than other children, and their speech is often difficult to understand. The disorder is believed to arise from impairments in brain regions, such as the striatum, that control the movements of the lips, mouth, and tongue. Foxp2 is also expressed in the brains of songbirds such as zebra finches and is critical to those birds’ ability to learn songs.
Foxp2 encodes a transcription factor, meaning that it can control the expression of many other target genes. Many species express Foxp2, but humans have a special form of Foxp2. In a 2014 study , Graybiel and colleagues found evidence that the human form of Foxp2, when expressed in mice, allowed the mice to accelerate the switch from declarative to procedural types of learning.
In that study, the researchers showed that mice engineered to express the human version of Foxp2, which differs from the mouse version by only two DNA base pairs, were much better at learning mazes and performing other tasks that require turning repeated actions into behavioral routines. Mice with human-like Foxp2 also had longer dendrites — the slender extensions that help neurons form synapses — in the striatum, which is involved in habit formation as well as motor control.
In the new study, the researchers wanted to explore how the Foxp2 mutation that has been linked with apraxia affects speech production, using ultrasonic vocalizations in mice as a proxy for speech. Many rodents and other animals such as bats produce these vocalizations to communicate with each other.
While previous studies, including the work by Liu and Graybiel in 2016, had suggested that Foxp2 affects dendrite growth and synapse formation, the mechanism for how that occurs was not known. In the new study, led by Liu, the researchers investigated one proposed mechanism, which is that Foxp2 affects motor proteins.
One of these molecular motors is the dynein protein complex, a large cluster of proteins that is responsible for shuttling molecules along microtubule scaffolds within cells.
“All kinds of molecules get shunted around to different places in our cells, and that's certainly true of neurons,” Graybiel says. “There’s an army of tiny molecules that move molecules around in the cytoplasm or put them into the membrane. In a neuron, they may send molecules from the cell body all the way down the axons.”
A delicate balance
The dynein complex is made up of several other proteins. The most important of these is a protein called dynactin1, which interacts with microtubules, enabling the dynein motor to move along microtubules. In the new study, the researchers found that dynactin1 is one of the major targets of the Foxp2 transcription factor.
The researchers focused on the striatum, one of the regions where Foxp2 is most often found, and showed that the mutated version of Foxp2 is unable to suppress dynactin1 production. Without that brake in place, cells generate too much dynactin1. This upsets the delicate balance of dynein-dynactin1, which prevents the dynein motor from moving along microtubules.
Those motors are needed to shuttle molecules that are necessary for dendrite growth and synapse formation on dendrites. With those molecules stranded in the cell body, neurons are unable to form synapses to generate the proper electrophysiological signals they need to make speech production possible.
Mice with the mutated version of Foxp2 had abnormal ultrasonic vocalizations, which typically have a frequency of around 22 to 50 kilohertz. The researchers showed that they could reverse these vocalization impairments and the deficits in the molecular motor activity, dendritic growth, and electrophysiological activity by turning down the gene that encodes dynactin1.
Mutations of Foxp2 can also contribute to autism spectrum disorders and Huntington’s disease, through mechanisms that Liu and Graybiel previously studied in their 2016 paper and that many other research groups are now exploring. Liu’s lab is also investigating the potential role of abnormal Foxp2 expression in the subthalamic nucleus of the brain as a possible factor in Parkinson’s disease.
The research was funded by the Ministry of Science and Technology of Taiwan, the Ministry of Education of Taiwan, the U.S. National Institute of Mental Health, the Saks Kavanaugh Foundation, the Kristin R. Pressman and Jessica J. Pourian ’13 Fund, and Stephen and Anne Kott.
Share this news article on:
Related links.
- Ann Graybiel
- McGovern Institute
- Department of Brain and Cognitive Sciences
Related Topics
- Brain and cognitive sciences
- Neuroscience
Related Articles

How Huntington’s disease affects different neurons

Neuroscientists get at the roots of pessimism

Distinctive brain pattern helps habits form

Neuroscientists identify key role of language gene
Previous item Next item
More MIT News

Women in STEM — A celebration of excellence and curiosity
Read full story →

A blueprint for making quantum computers easier to program

“Nanostitches” enable lighter and tougher composite materials

From neurons to learning and memory

A biomedical engineer pivots from human movement to women’s health

MIT tops among single-campus universities in US patents granted
- More news on MIT News homepage →
Massachusetts Institute of Technology 77 Massachusetts Avenue, Cambridge, MA, USA
- Map (opens in new window)
- Events (opens in new window)
- People (opens in new window)
- Careers (opens in new window)
- Accessibility
- Social Media Hub
- MIT on Facebook
- MIT on YouTube
- MIT on Instagram
- Published: 18 April 2013
Neurogenomics of speech and language disorders: the road ahead
- Pelagia Deriziotis 1 &
- Simon E Fisher 1 , 2
Genome Biology volume 14 , Article number: 204 ( 2013 ) Cite this article
10k Accesses
25 Citations
8 Altmetric
Metrics details
Next-generation sequencing is set to transform the discovery of genes underlying neurodevelopmental disorders, and so offer important insights into the biological bases of spoken language. Success will depend on functional assessments in neuronal cell lines, animal models and humans themselves.
Introduction
The human capacity for complex spoken language is unique [ 1 ]. Speech endows us with the ability to verbally express our ideas, opinions and feelings, using rapid precise control of the oral motor structures (larynx, mouth, tongue) to convert our thoughts into streams of sound that can be decoded by others. While vocal communication in other species sometimes exploits simple mappings between sound and meaning, the reach of human language extends far beyond this, most notably through its extraordinary generative power. A discrete number of individual units of language can be combined into a limitless number of utterances, giving us the potential to express and comprehend an infinite array of concepts. Moreover, when growing up in a language-rich environment, any normal human infant becomes highly proficient in his or her native language with astonishing ease, and without the need for explicit teaching.
It has been argued for many years that inherited factors must make a key contribution to the acquisition of spoken language [ 2 ]. It is only in the past decade or so, with the rise of molecular genetics, that biologists have been able to provide the first robust empirical evidence regarding this issue. To begin investigating the pathways involved, research has focused on the roles of genes, proteins and cellular machinery in the etiology of language impairments, in which people mysteriously fail to develop normal skills despite adequate linguistic input and opportunity [ 3 ]. There is a diverse array of these language-related disorders, which usually appear in early childhood and often persist into later life, and they are common enough to have a major impact on modern society. Language problems are frequently observed co-occurring with other developmental disorders, such as autism and epilepsy [ 4 , 5 ].
Prior to the advent of molecular studies of language disorders, the importance of the genome was already evident from epidemiological analyses. These disorders typically cluster in families [ 6 – 9 ] and monozygotic twins display substantially higher rates of concordance than dizygotic twins [ 10 – 12 ]. Clearly, acquisition of fluent spoken language is also influenced by the environment and its interaction with our genes. However, beyond the obvious effects of impoverished language input (for example, due to hearing problems) there is little known regarding specific environmental risk factors that may disturb linguistic development [ 13 ].
Initial clues to the molecular bases of speech and language impairments came from low-density linkage screens [ 14 ], followed by targeted association studies of particular chromosome regions and/or focused mutation screens of candidate genes [ 15 ]. In addition, studies of chromosomal abnormalities are contributing to our understanding of such disorders, and genome-wide association scans using hundreds of thousands of single nucleotide polymorphisms (SNPs) are underway in several cohorts. However, it is evident that the future of gene discovery in language-related traits, as for many other complex phenotypes, lies in large-scale DNA sequencing of entire human genomes.
Traditional sequencing methods are slow, laborious and expensive; the original human genome sequencing project cost more than US$3 billion and took more than a decade to finish [ 16 ]. Dramatic technological advances have transformed the ability to analyze our genetic makeup at single nucleotide resolution and commercialization of these 'next-generation' platforms is growing fast. At the time of writing, a human genome can be entirely sequenced in a matter of days for only a few thousand dollars, and costs continue to fall at a remarkable rate. Nevertheless, excitement over the enormous potential of the new technologies must be tempered by acknowledging the associated analytical challenges. Already, our capacity to rapidly generate large swathes of sequence data from many individuals outstrips our capacity to infer the underlying biology of a trait using such information.
Here, we begin by summarizing approaches previously applied to identify and study the first genes implicated in speech and language disorders (Table 1 ). We go on to discuss the promise of next-generation sequencing (NGS) for uncovering the key genomic changes that affect our speech and language abilities, not only in relevant disorders, but also in the general population. We argue that it is essential to be able to assess the functional significance of identified variants if we are to understand their biological impact and elucidate their contributions to the human traits of interest. The success of such efforts will depend on synergies between diverse research techniques, including bioinformatics and experimental analyses using model systems, as well as integration of human genome sequences and functional gene network datasets (Figure 1 ).

Neurogenomics of speech and language disorders . Next-generation sequencing will yield large datasets of genomic variants with potential relevance for speech and language. Identification of key variants is critically dependent on multidisciplinary studies of function in cell lines, animal models and humans, along with integration of data on neurogenetic networks, as detailed in the text. The image under 'Next-generation sequencing' comes from istockphoto.com (DNA code; File #9614920), the boxshade plot under ' In silico analyses' is a subpart taken from Figure 4 of [ 17 ], the lefthand bottom panel of 'Cellular assays' is a subpart taken from Supplementary Figure 5c of [ 68 ], the 'Neurogenetic networks' image is taken from Figure 4b of [ 82 ] and the Zebrafinch image is reproduced with permission from Geoffrey Dabb and Canberra Ornithologists Group.
Gene mapping in speech and language disorders
Speech apraxia.
The first gene to be clearly implicated in a speech and language disorder was FOXP2 . Disruptions of this gene cause a monogenic form of developmental verbal dyspraxia (DVD), also known as childhood apraxia of speech (CAS) [ 17 ], characterized by problems with the learning and execution of coordinated movement sequences of the mouth, tongue, lips and soft palate [ 18 , 19 ]. FOXP2 was discovered through molecular studies of a large three-generational pedigree (the KE family) in which half the members have CAS, accompanied by wide-ranging deficits in both oral and written language, affecting not only production but also comprehension [ 17 ]. Linkage mapping in this family identified a region on chromosome 7q31 that co-segregated perfectly with the disorder [ 20 ]. An unrelated child with similar speech and language deficits was found to carry a de novo balanced translocation involving the same interval, which directly interrupted the coding region of a novel gene, FOXP2 [ 17 , 21 ]. Screening of FOXP2 in the KE family revealed that all affected members had inherited a heterozygous point mutation yielding an amino acid substitution at a key residue of the encoded protein [ 17 ]. Subsequent studies identified additional etiological FOXP2 variants (nonsense mutations, translocations, deletions) in individuals and families with speech and language problems, typically including CAS as a core feature (reviewed by Fisher and Scharff [ 22 ]). Although etiological mutations of FOXP2 are rare [ 23 , 24 ], the gene provides a valuable molecular window into neurogenetic mechanisms contributing to human spoken language, as detailed elsewhere in this article.
Beyond FOXP2 , additional loci that may contribute to CAS have emerged from cases of chromosomal abnormalities, identified using cytogenetic screening and/or comparative genomic hybridization (CGH). One report described a family in which three affected siblings all carry an unbalanced 4q;16q translocation [ 25 ]. Another study defined a small region on 12p13.3, containing the ELKS/ERC1 gene, commonly deleted in nine unrelated patients with delayed speech development, most of whom had a formal diagnosis of CAS [ 26 ]. Interestingly, a key isoform encoded by ELKS/ERC1 appears to be expressed specifically in the brain, where it binds to RIM proteins. In neurons, RIMs act within the presynaptic active zone, a site that integrates synaptic vesicle exo/endocytosis with intracellular signaling in the nerve terminal [ 27 ]. Certain copy number variant (CNV) syndromes with complex variable phenotypes have been linked to increased risk of CAS, including 16p11.2 microdeletions [ 28 , 29 ] and 7q11.23 microduplications [ 30 ]. The rare metabolic disorder, galactosemia, is also associated with elevated incidence of CAS [ 31 ].
Specific language impairment
When a child is delayed or impaired in acquiring language, without any obvious physical or neurological cause (cleft lip/palate, intellectual disability (ID), autism, deafness, and so on) he or she is usually diagnosed with specific language impairment (SLI). Since it is defined using exclusionary criteria, SLI encompasses a range of different cognitive and behavioral profiles. The most common forms involve deficits in expressive language, either in isolation or accompanied by receptive problems.
The estimated prevalence of SLI is up to 7% in kindergarten children [ 32 ] and it shows familial clustering; twin studies consistently indicate high heritability [ 10 , 11 , 33 ]. In contrast to the rare cases of monogenic CAS discussed above, typical forms of SLI have a complex multifactorial basis [ 34 ]. Genome-wide linkage mapping in families with SLI have suggested the existence of multiple risk loci, on chromosomes 16q and 19q [ 35 – 38 ], as well as 2p and 13q [ 39 , 40 ]. Targeted analysis of 16q identified variants in two genes, ATP2C2 and CMIP , associated with deficits on a non-word repetition task, considered to be an index of impaired phonological short-term memory [ 15 , 41 ]. The ATP2C2 gene encodes a single subunit integral membrane P-type ATPase that catalyzes the ATP-driven transport of cytosolic calcium and manganese into the Golgi lumen [ 42 ]. This cellular role makes it a plausible candidate for SLI susceptibility, since intracellular calcium levels are intimately linked to multiple diverse aspects of neuronal function, ranging from migration to plasticity, while manganese dysregulation has been linked to neurodegenerative phenotypes. The product of CMIP contains pleckstrin homology and leucine-rich repeat domains, and is hypothesized to be an adaptor protein of the actin cytoskeleton, interacting with filamin A and RelA (an NF-kappaB subunit) [ 43 ]. Although little is known about CMIP at this stage, it is again a credible candidate for involvement in nervous system function, since cytoskeletal reorganization makes essential contributions to processes like neuronal migration and synapse formation/modification. Other candidate genes (such as CNTNAP2 ) have been implicated in SLI susceptibility through functional approaches [ 44 ], as highlighted elsewhere in this article.
Studies of isolated founder populations may also help pinpoint new genes contributing to language disorders. A notable example is Robinson Crusoe Island - an island of 633 residents lying west of Chile, South America - which was most recently colonized in the late 19th century [ 45 ]. Thirty-five percent of the colonizing children satisfy criteria for a diagnosis of SLI, substantially higher than the 4% prevalence rate for mainland Chile [ 45 ]. Initial molecular investigations identified several genomic regions of interest (on chromosomes 6, 7, 12, 13 and 17), but no specific risk genes have yet been discovered [ 46 ].
SLI has connections with another heritable neurodevelopmental trait, dyslexia, defined as specific significant impairments in reading and/or spelling that are not attributed to intelligence, visual acuity problems or inadequate learning opportunities. Although they do not display overt difficulties with speech or language, people with dyslexia often have subtle underlying deficits with aspects of linguistic processing [ 47 ]. Thus, genetic studies of dyslexia may be informative for understanding language pathways. We do not have space to discuss this here, and refer readers to other recent reviews [ 48 , 49 ].
Stuttering is a neurodevelopmental disorder that disturbs the flow of speech [ 50 ]. People who stutter are affected by uncontrollable repetitions and prolongations of syllables, and by involuntary silent pauses while speaking; these difficulties begin in childhood, persisting in about 20% of case referrals [ 51 ]. Most people who suffer from persistent stuttering nevertheless display normal linguistic proficiency [ 52 ]. Stuttering is thought to have a strong genetic basis [ 53 ]. Thus far, most genome-wide investigations of persistent familial stuttering have revealed only suggestive evidence of linkage, with loci distributed across at least ten chromosomes, and little overlap between different studies, indicating that this is a complex multifactorial trait [ 53 – 55 ].
One of the few reports of significant linkage focused on 46 consanguineous families from Pakistan, and highlighted chromosome 12q as a site of interest [ 56 ]. Subsequent analyses of the largest family from that study found that most affected relatives carried a coding variant in the 12q23.2 gene GNPTAB , which encodes two subunits of GlcNAc-phosphotransferase (GNPT) [ 57 ]. This putative risk variant (Q1200K), which altered a conserved residue of the protein, was identified in a number of other Pakistani cases, at higher frequency than Pakistani controls. GNPT is involved in addition of a mannose 6-phosphate tag to hydrolytic enzymes, allowing them to be targeted to lysosomes. Further screening of GNPTAB , as well as GNPTG and NAGPA , two closely related genes in this metabolic pathway, identified several different coding variants that were only present in cases and not controls [ 57 ]. The proposed risk variants are rare even among people who stutter, so it is likely that there are other unknown genes involved in stuttering.
The next generation: uncovering novel risk variants
While it is clear that exciting progress has been made, many of the genetic risk factors underlying speech and language disorders and/or normal linguistic variation remain to be discovered. At the time of writing, no study had yet reported the use of NGS methodologies to specifically investigate language-related traits. However, the advent of NGS has transformed the identification of genetic variants in other important neurodevelopmental phenotypes that co-occur with language deficits, such as ID and autism spectrum disorders (ASDs). Thus far, most such research has focused on sequencing protein-coding regions of the genome (the exome) to detect de novo variants in rare and common forms of these disorders [ 58 – 60 ]. Since de novo mutations have highly deleterious effects and are subject to strong negative selection, it is hypothesized that they might be important explanations of sporadic occurrences of disorder.
Whole-exome sequencing first proved effective in detecting causal de novo variants in rare reproductively lethal neurodevelopmental disorders, such as Kabuki syndrome [ 61 ], Bohring-Opitz syndrome [ 62 ] and KBG syndrome [ 63 ]. The study that pioneered this approach assessed 13 cases of Schinzel-Giedion syndrome, which is characterized by severe ID and typical facial features, and revealed de novo gain-of-function mutations independently occurring in a single gene, SETBP1 [ 64 ]. Interestingly, haploinsufficiency of SETBP1 has been identified in some cases of expressive speech impairment [ 65 ]. SETBP1 encodes a widely expressed nuclear protein that interacts with SET, an oncogene involved in DNA replication. Recent studies have shown that SET binding protein 1 (SETBP1) also includes three highly conserved AT-hooks (motifs that bind AT-rich DNA in a non-sequence-specific manner) and that it can act as a transcription factor, directly activating targets such as Hoxa9 and Hoxa10 [ 66 ]. Functional links between SETBP1 and brain development have yet to be explored.
NGS techniques are also shedding light on the roles of de novo changes in common non-syndromic disorders [ 59 ]. A pilot study of whole-exome sequencing in sporadic cases of non-syndromic ID and their parents (parent-child trios) reported nine non-synonymous de novo mutations in different genes in seven of ten probands [ 67 ]. Since then, multiple investigations have employed similar approaches to screen trios or quads (trio plus unaffected siblings), including four large-scale whole-exome sequencing efforts across about 1,000 ASD families [ 68 – 72 ] (reviewed by Buxbaum et al. [ 60 ]). One conclusion of this work was that the rate of de novo mutations was higher in ASD probands than controls, and it pointed to six genes of particular interest that had recurrent loss-of-function mutations.
A major advantage of focusing on de novo mutations is that it dramatically reduces the search space for potential causative variants; it is estimated that an average of approximately one de novo coding variant arises per genome per generation [ 59 ]. Interpretation of NGS data becomes more difficult when the search criteria are broadened to encompass all potential etiological coding variants that a proband carries, and it is even more challenging if one also considers non-exonic variations throughout the entire genome. It is not currently known if the genetic architecture underlying specific speech and language disorders includes a significant role for de novo mutations. Thus, it will be important to develop alternative study designs and analytic strategies (for example, Yu et al. [ 73 ] and Lim et al. [ 74 ]) for pinpointing causative mutations in NGS data from cases and families with language impairments.
Bridging the gap from genetic variants to biology
In the near future, NGS methods will become standard tools in molecular studies of speech and language disorders. As noted above, gene discovery strategies will need to move beyond the de novo paradigms that have been so successful for ID and ASD. Researchers will be faced with the major challenge of discerning which of the many plausibly causal variants carried by each affected person are physiologically relevant to their speech and/or language impairments. Fortunately, distinct fields combining computational and experimental methods can help ascertain the biological roles of detected variants and ultimately highlight genes important for our unique capacity for spoken language.
When focusing on protein-coding sequences, after initial filtering of identified variants from NGS data, it is possible to use predictive algorithms such as SIFT [ 75 ] and PolyPhen2 [ 76 ] to flag the most promising mutations for subsequent analyses. Computational methods such as these use known information on protein sequence and evolutionary history to rank them as benign, possibly damaging or probably damaging. Nonetheless, as cellular pathways harbor some degree of redundancy, not all loss-of-function mutations will contribute to a given disorder and such predictions should be treated with caution. For example, sequencing of FOXP2 in a cohort of CAS/DVD cases revealed a non-synonymous substitution near the N-terminus of the protein (Q17L) in one of the probands [ 24 ], a variant that is predicted to be damaging by both SIFT and PolyPhen2. However, follow-up functional experiments of the Q17L substitution using cell models did not find adverse effects on protein characteristics, in contrast to observations for other proband mutations [ 77 ]. Together with the fact that the Q17L proband has an affected sibling who does not carry the substitution, it seems unlikely that this particular change is etiological. Thus, although bioinformatic approaches help narrow down the list of variants from ongoing high-throughput genetic screens of speech and language phenotypes, experimental analyses in model systems are often crucial for determining causality, as well as offering deeper insights into mechanisms.
The value of functional approaches is particularly apparent from studies of how FOXP2 mutations lead to speech and language disorder [ 22 ]. FOXP2 encodes a forkhead-box transcription factor. Following homo- or hetero-dimerization with other forkhead box P (FOXP) family members [ 78 ], the protein binds DNA and represses transcription of its target genes [ 79 ]. Human neuron-like cells have been used to assess two different mutant FOXP2 proteins that co-segregate with disorder in CAS/DVD families: pFOXP2.R553H [ 17 ] and pFOXP2.R328X [ 24 ]. The functional assays demonstrated that these mutations severely disrupt nuclear localization, DNA-binding ability and transactivation potential of the protein [ 77 ]. Investigations into downstream targets of FOXP2 highlighted several neuronal pathways that it regulates. Independent high-throughput studies of promoter occupancy in cells and human fetal brain reported that FOXP2 directly regulates genes involved in neurite outgrowth, synaptic plasticity and axon guidance [ 80 , 81 ]. More recently, following genome-wide analyses of neural targets in vivo in mouse models, it has been shown that Foxp2 mutations can alter neurite outgrowth and branching in primary neurons [ 82 ].
A subset of FOXP2 targets are implicated in neurodevelopmental disorders that often co-occur with language deficits, such as the sushi repeat-containing protein X-linked 2 (SRPX2)-plasminogen activator receptor, urokinase-type (uPAR) complex in epilepsy and speech apraxia [ 83 ], DISC1 in schizophrenia [ 84 ] and MET in ASD [ 85 ]. The most rigorously studied FOXP2 target is CNTNAP2 , encoding contactin-associated protein-like 2 (CASPR2), a transmembrane scaffolding protein that clusters K + channels in myelinated axons [ 86 ]. CASPR2 is a member of the neurexin superfamily and, in addition to its role in mature neurons, it has been implicated in neuronal migration, dendritic arborization and spine development [ 87 ]. Homozygous loss-of-function CNTNAP2 mutations cause infant-onset epilepsy, learning deficits and language regression [ 88 ]. FOXP2 binds directly within the first intron of CNTNAP2 and is able to downregulate its expression [ 44 ]. Association analyses of quantitative phenotype data in 184 small SLI families identified a cluster of common intronic SNPs in CNTNAP2 that correlated significantly with reduced performance on linguistic tests, most strongly for the non-word repetition endophenotype [ 44 ]. The identity of the precise functional variant(s) in this region is not yet determined, but it is hypothesized that they affect the way that CNTNAP2 is regulated. Rare and common CNTNAP2 variants have also been implicated independently in ASDs [ 89 – 91 ], consistent with prior hypotheses that SLI and ASDs may involve some degree of shared genetic etiology. Beyond SLI, ASD and epilepsy, contributions of CNTNAP2 have been suggested for a range of other neurodevelopmental phenotypes, including schizophrenia [ 92 ], selective mutism [ 93 ] and Tourette syndrome [ 94 ].
A recent study of sporadic ASD demonstrates how the combination of NGS screens with functional experiments can shed light on language-related gene networks [ 68 ]. Whole-exome sequencing of parent-child trios identified a de novo frameshift mutation in an ASD proband, introducing a premature stop codon in FOXP1 [ 68 ]. The child was severely affected, with regression and language delays. FOXP1 is the most closely related gene to FOXP2 in the human genome and they can act synergistically to regulate shared targets in regions of co-expression [ 78 , 95 , 96 ]. Remarkably, the proband with the FOXP1 mutation also carried an extremely rare CNTNAP2 missense variant, inherited from his unaffected mother [ 68 ]. In cell-based functional analyses, the aberrant FOXP1 protein mislocalized to the cytoplasm and lost its transcriptional repressor properties; expression of the mutant FOXP1 isoform in cells elevated CNTNAP2 levels, unlike wild-type FOXP1 [ 68 ]. These data were consistent with a two-hit mechanism in which abnormal FOXP1 results in higher CNTNAP2 levels, amplifying any potentially deleterious effects of the missense CNTNAP2 variant of the proband [ 68 ]. Similar findings regarding multiple-hit mechanisms have emerged from independent studies of ASDs and other neurodevelopmental syndromes (for example, Leblond et al. [ 97 ]), suggesting that this may be an important model for genetic etiology of such disorders [ 98 ].
Previous screening of 49 children diagnosed with CAS/DVD did not detect any obviously etiological FOXP1 mutations [ 99 ]. However, studies of patients with mild to moderate ID and language impairment have detected rare de novo deletions and a nonsense FOXP1 variant [ 100 , 101 ]. High-throughput sequencing of balanced chromosomal abnormalities in neurodevelopmental disorders identified disruptions at the FOXP1 locus [ 102 ].
There has been little reported to date on functional analyses of other genes (such as ATP2C2 and CMIP ) associated with speech and language disorders, in part because no protein-coding variants have been pinpointed. As noted above, some cases of persistent stuttering carry coding variants in genes ( GNPTAB , GNPTG and NAGPA ) involved in lysosomal targeting of hydrolase enzymes. Interestingly, loss-of-function mutations of this pathway cause mucolipidosis disorders, which involve severe abnormalities affecting multiple systems, including skeletal, respiratory and cardiovascular tissues. Cell-based assays were recently used to analyze Mannose 6-phosphate-uncovering enzyme variants found in people who stutter, and were reported to yield incorrect protein folding, decreased enzymatic activity and degradation by the proteasome [ 103 ].
It is not always feasible to carry out experimental assessments of putative risk variants. The nature of assessment is highly dependent on the type of gene product; it is difficult to test protein function if there are no known measurable properties. In contrast to NGS technologies, functional experiments typically remain high cost, time-consuming and laborious, and are less amenable for upscaling. Nevertheless, as NGS reveals additional variants potentially implicated in language impairments and other neurodevelopmental traits, we will inevitably need access to high-throughput techniques for simultaneous mutation testing to define disease-causing variants across the genome [ 104 ]. Indeed, several multiplex approaches for characterizing the functional effects of genetic variation in proteins [ 105 ], mammalian regulatory elements [ 106 , 107 ] and RNA [ 108 ] have recently been developed. More and more emphasis will be placed on possible functional variants that lie outside protein-coding regions. Various efforts are underway to facilitate this transition, most notably the ENCODE project, which aims to characterize all functional elements at a genome-wide scale, including non-coding RNA and cis -regulatory elements [ 109 ]. RegulomeDB is of particular interest, as it combines data from the ENCODE project, GEO and published literature into a single, integrated database that can be used to query the functional significance of variants in both coding and non-coding regions of the genome [ 110 ].
Integrating data networks
Beyond establishing causality, functional characterization of candidate risk variants in model organisms may also help highlight pathways implicated in the origins and bases of language. For example, studies of FOXP2 across different species (mouse, bird, human) have given us initial clues into neurogenetic networks facilitating human spoken language [ 22 , 111 ]. FOXP2 expression is enriched in several brain areas, including the basal ganglia, deep cortical layers, thalamus and cerebellum [ 112 ], some of which display subtle structural and functional abnormalities in people carrying FOXP2 mutations [ 19 , 112 – 114 ]. From an evolutionary perspective, this is a highly conserved gene with regard to both the amino acid sequence of the encoded protein and the neural sites where it is expressed [ 95 , 115 ]. These data suggest that ancestral forms of FOXP2 were involved in important aspects of brain development long before the emergence of spoken language. There is evidence that the functions of the gene may have been modified during human evolution ([ 116 ]; also see below), but it remains clear that its roles in the human brain are built on evolutionarily ancient pathways [ 1 ].
Extensive characterization of rodent models carrying etiological Foxp2 variants indicates roles in synaptic plasticity, motor-skill learning, and processing and integration of auditory information [ 117 – 120 ]. When mice are heterozygous for the mutation that causes speech problems in the human KE family, they display decreased synaptic plasticity in corticostriatal circuits and motor-skill learning deficits [ 117 ]. These mouse findings are intriguing given that affected humans have problems learning to master the rapid coordinated orofacial movements underlying speech [ 121 ]. In vivo electrophysiology recordings in awake-behaving mice revealed more about the impacts of Foxp2 on corticostriatal circuitry; mice heterozygous for the KE mutation displayed higher basal striatal activity than wild-type controls, and medium spiny neurons showed aberrant negative modulation of their firing rates during motor-skill learning [ 118 ]. Separate studies used mouse models to explore whether impairments in auditory processing and auditory-motor integration might also be relevant to FOXP2 -related disorders [ 119 , 120 ]. Mice carrying the KE mutation were reported to have altered auditory brainstem responses to sound, although this finding was not replicated in mice carrying a different mutation associated with speech/language problems in another family [ 119 ]. Mice carrying either etiological mutation have deficits in learning to associate auditory stimuli with motor outputs [ 120 ].
Songbirds carry their own version of FOXP2 , referred to as FoxP2 , and it appears to make important contributions to the functions of a striatal nucleus called Area X [ 122 ]. In zebra finches, Area X is critical for auditory-guided vocal learning, a process in which young male birds learn their song by imitating an adult tutor. Vocal learning is also a key component of human speech acquisition. FoxP2 mRNA levels in Area X are enriched in young birds during the critical song-learning period [ 123 ] and show rapid downregulation when adult birds practice their songs outside the context of courtship [ 124 – 126 ]. Furthermore, selective knockdown of FoxP2 in Area X disrupts the song-learning process [ 127 ] and alters dendritic spine density in this region [ 128 ].
Functional studies of genes implicated in language-related disorders may also give us entry points into mechanisms involved in language function in the general population. As discussed above, variants of CNTNAP2 , a direct target of FOXP2, were associated with linguistic deficits in clinically distinct neurodevelopmental disorders [ 44 , 88 , 89 , 129 – 131 ]. Subsequent studies revealed that CNTNAP2 may contribute to language processing in healthy individuals [ 132 – 134 ]. The cluster of CNTNAP2 SNPs that is associated with language phenotypes in SLI and ASDs has also been reported to correlate with assessments of early language development in general population samples [ 132 ]. Neuroimaging genetics studies of common CNTNAP2 SNPs in healthy samples have proposed associations with functional brain measures related to language [ 133 , 134 ] and with altered structural connectivity patterns [ 135 ]. However, imaging genetics of language is a field that is only in its infancy; reports thus far involved small sample sizes with limited power, as well as a substantial multiple-testing burden, and results of different studies have been largely inconsistent. Additional analyses are required to elucidate how FOXP2 , CNTNAP2 and other language-related genes influence brain circuits at multiple levels of description - molecular, cellular, structural and functional.
Insights from ancient genomes
The reach of NGS technologies extends well beyond living species. These innovations have allowed molecular anthropologists to reconstruct large portions of nuclear genomes from extinct hominins that co-existed with our ancestors, such as Neanderthals [ 136 ] and Denisovans [ 137 ]. By comparing modern human sequences to ancient hominin genomes, as well as to our closest extant relatives, chimpanzees, it is possible to identify molecular variants that arose during human evolution, and roughly date them with regard to branches of the primate phylogenetic tree. As for other NGS projects, our capacity to generate large amounts of sequence data exceeds our ability to interpret it. So although scientists have successfully catalogued many of the DNA changes that occurred on our lineage, an extraordinary feat in itself, it is still a major challenge to determine which of these evolutionary events were relevant for the emergence of traits such as speech and language acquisition [ 1 ]. Here, success may depend on the integration of findings from evolutionary genomics with data from molecular studies of language-related disorders.
The best illustration of this approach comes again from work on the FOXP2 gene, which was targeted for evolutionary investigations, based on its prior link to a severe speech and language disorder. Comparative primate genomics suggests that FOXP2 probably underwent at least two interesting evolutionary events on the lineage that led to modern humans. After splitting from the chimpanzee (several million years ago) there were changes in the coding region of the locus that yielded two amino acid substitutions in the encoded protein [ 138 ]. Although these are minor changes outside the known functional domains, when such substitutions are inserted into the endogenous Foxp2 gene of a mouse, they have subtle detectable effects on brain structure and function, including altered connectivity and plasticity of corticostriatal circuits [ 116 ]. NGS approaches indicate that these amino acid substitutions are shared by Neanderthals [ 136 ] and Denisovans [ 137 ]. (It is worth emphasizing here that status of a single gene is not enough to determine whether or not a species can speak.) Researchers went on to identify a number of non-coding variants in intronic regions of FOXP2 that had occurred more recently on the human lineage, after splitting from Neanderthal/Denisovan a few hundred thousand years ago [ 139 ]. One of these changes lies in a region that underwent a recent selective sweep, and alters a putative binding site for the POU class 3 homeobox 2 (POU3F2) transcription factor, such that it may have affected regulation of FOXP2 expression; cell-based analyses are consistent with this hypothesis [ 139 ]. Thus, just like sequence-based analyses of language-related disorders, evaluation of the biological significance of interesting variants from ancient genomics requires functional studies using model systems.
The advent of whole genome NGS means that data generation will no longer be the limiting factor in understanding how genetic factors contribute to mechanisms underlying complex neurodevelopmental traits. Coupling NGS approaches to functional validation in model systems will facilitate network mapping and pathway investigation in speech and language disorders, and ultimately in normal linguistic development.
Abbreviations
autism spectrum disorder
childhood apraxia of speech
developmental verbal dyspraxia
forkhead box P
intellectual disability
- next-generation sequencing
specific language impairment.
Fisher SE, Marcus GF: The eloquent ape: genes, brains and the evolution of language. Nat Rev Genet. 2006, 7: 9-20.
PubMed CAS Google Scholar
Pinker S, Bloom P: Natural language and natural selection. Behav Brain Sci. 1990, 13: 707-784. 10.1017/S0140525X00081061.
Google Scholar
Bishop DV: Genes, cognition, and communication: insights from neurodevelopmental disorders. Ann N Y Acad Sci. 2009, 1156: 1-18. 10.1111/j.1749-6632.2009.04419.x.
PubMed CAS PubMed Central Google Scholar
Bishop DV: Overlaps between autism and language impairment: phenomimicry or shared etiology?. Behav Genet. 2010, 40: 618-629. 10.1007/s10519-010-9381-x.
Pal DK: Epilepsy and neurodevelopmental disorders of language. Curr Opin Neurol. 2011, 24: 126-131. 10.1097/WCO.0b013e328344634a.
PubMed Google Scholar
Neils J, Aram DM: Family history of children with developmental language disorders. Percept Mot Skills. 1986, 63: 655-658. 10.2466/pms.1986.63.2.655.
Tomblin JB: Familial concentration of developmental language impairment. J Speech Hear Disord. 1989, 54: 287-295.
Barry JG, Yasin I, Bishop DV: Heritable risk factors associated with language impairments. Genes Brain Behav. 2007, 6: 66-76. 10.1111/j.1601-183X.2006.00232.x.
Lewis BA, Freebairn LA, Hansen AJ, Miscimarra L, Iyengar SK, Taylor HG: Speech and language skills of parents of children with speech sound disorders. Am J Speech Lang Pathol. 2007, 16: 108-118. 10.1044/1058-0360(2007/015).
Lewis BA, Thompson LA: A study of developmental speech and language disorders in twins. J Speech Hear Res. 1992, 35: 1086-1094.
Bishop DV, North T, Donlan C: Genetic basis of specific language impairment: evidence from a twin study. Dev Med Child Neurol. 1995, 37: 56-71.
Bishop DV, Hayiou-Thomas ME: Heritability of specific language impairment depends on diagnostic criteria. Genes Brain Behav. 2008, 7: 365-372. 10.1111/j.1601-183X.2007.00360.x.
Bishop DV: Genetic and environmental risks for specific language impairment in children. Philos Trans R Soc Lond B Biol Sci. 2001, 356: 369-380. 10.1098/rstb.2000.0770.
Fisher SE, Lai CS, Monaco AP: Deciphering the genetic basis of speech and language disorders. Annu Rev Neurosci. 2003, 26: 57-80. 10.1146/annurev.neuro.26.041002.131144.
Newbury DF, Monaco AP: Genetic advances in the study of speech and language disorders. Neuron. 2010, 68: 309-320. 10.1016/j.neuron.2010.10.001.
International Human Genome Sequencing Consortium: Finishing the euchromatic sequence of the human genome. Nature. 2004, 431: 931-945. 10.1038/nature03001.
Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP: A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001, 413: 519-523. 10.1038/35097076.
Alcock KJ, Passingham RE, Watkins KE, Vargha-Khadem F: Oral dyspraxia in inherited speech and language impairment and acquired dysphasia. Brain Lang. 2000, 75: 17-33. 10.1006/brln.2000.2322.
Watkins KE, Vargha-Khadem F, Ashburner J, Passingham RE, Connelly A, Friston KJ, Frackowiak RSJ, Mishkin M, Gadian DG: MRI analysis of an inherited speech and language disorder: structural brain abnormalities. Brain. 2002, 125: 465-478. 10.1093/brain/awf057.
Fisher SE, Vargha-Khadem F, Watkins KE, Monaco AP, Pembrey ME: Localisation of a gene implicated in a severe speech and language disorder. Nat Genet. 1998, 18: 168-170. 10.1038/ng0298-168.
Lai CS, Fisher SE, Hurst JA, Levy ER, Hodgson S, Fox M, Jeremiah S, Povey S, Jamison DC, Green ED, Vargha-Khadem F, Monaco AP: The SPCH1 region on human 7q31: genomic characterization of the critical interval and localization of translocations associated with speech and language disorder. Am J Hum Genet. 2000, 67: 357-368. 10.1086/303011.
Fisher SE, Scharff C: FOXP2 as a molecular window into speech and language. Trends Genet. 2009, 25: 166-177. 10.1016/j.tig.2009.03.002.
Newbury DF, Bonora E, Lamb JA, Fisher SE, Lai CS, Baird G, Jannoun L, Slonims V, Stott CM, Merricks MJ, Bolton PF, Bailey AJ, Monaco AP, International Molecular Genetic Study of Autism Consortium: FOXP2 is not a major susceptibility gene for autism or specific language impairment. Am J Hum Genet. 2002, 70: 1318-1327. 10.1086/339931.
MacDermot KD, Bonora E, Sykes N, Coupe AM, Lai CS, Vernes SC, Vargha-Khadem F, McKenzie F, Smith RL, Monaco AP, Fisher SE: Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits. Am J Hum Genet. 2005, 76: 1074-1080. 10.1086/430841.
Shriberg LD, Jakielski KJ, El-Shanti H: Breakpoint localization using array-CGH in three siblings with an unbalanced 4q;16q translocation and childhood apraxia of speech (CAS). Am J Med Genet. 2008, 146A: 2227-2233. 10.1002/ajmg.a.32363.
Thevenon J, Callier P, Andrieux J, Delobel B, David A, Sukno S, Minot D, Mosca Anne L, Marle N, Sanlaville D, Bonnet M, Masurel-Paulet A, Levy F, Gaunt L, Farrell S, Le Caignec C, Toutain A, Carmignac V, Mugneret F, Clayton-Smith J, Thauvin-Robinet C, Faivre L: 12p13.33 microdeletion including ELKS/ERC1, a new locus associated with childhood apraxia of speech. Eur J Hum Genet. 2013, 21: 82-88. 10.1038/ejhg.2012.116.
Wang Y, Liu X, Biederer T, Sudhof TC: A family of RIM-binding proteins regulated by alternative splicing: Implications for the genesis of synaptic active zones. Proc Natl Acad Sci USA. 2002, 99: 14464-14469. 10.1073/pnas.182532999.
Newbury DF, Mari F, Sadighi Akha E, Macdermot KD, Canitano R, Monaco AP, Taylor JC, Renieri A, Fisher SE, Knight SJ: Dual copy number variants involving 16p11 and 6q22 in a case of childhood apraxia of speech and pervasive developmental disorder. Eur J Hum Genet. 2012, 21: 361-365.
PubMed PubMed Central Google Scholar
Raca G, Baas BS, Kirmani S, Laffin JJ, Jackson CA, Strand EA, Jakielski KJ, Shriberg LD: Childhood Apraxia of Speech (CAS) in two patients with 16p11.2 microdeletion syndrome. Eur J Hum Genet. 2013, 21: 455-459. 10.1038/ejhg.2012.165.
Van der Aa N, Rooms L, Vandeweyer G, van den Ende J, Reyniers E, Fichera M, Romano C, Delle Chiaie B, Mortier G, Menten B, Destrée A, Maystadt I, Männik K, Kurg A, Reimand T, McMullan D, Oley C, Brueton L, Bongers EM, van Bon BW, Pfund R, Jacquemont S, Ferrarini A, Martinet D, Schrander-Stumpel C, Stegmann AP, Frints SG, de Vries BB, Ceulemans B, Kooy RF: Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur J Med Genet. 2009, 52: 94-100. 10.1016/j.ejmg.2009.02.006.
Shriberg LD, Potter NL, Strand EA: Prevalence and phenotype of childhood apraxia of speech in youth with galactosemia. J Speech Lang Hear Res. 2011, 54: 487-519. 10.1044/1092-4388(2010/10-0068).
Tomblin JB, Records NL, Buckwalter P, Zhang X, Smith E, O'Brien M: Prevalence of specific language impairment in kindergarten children. J Speech Lang Hear Res. 1997, 40: 1245-1260.
Tomblin JB, Buckwalter PR: Heritability of poor language achievement among twins. J Speech Lang Hear Res. 1998, 41: 188-199.
Newbury DF, Bishop DV, Monaco AP: Genetic influences on language impairment and phonological short-term memory. Trends Cogn Sci. 2005, 9: 528-534. 10.1016/j.tics.2005.09.002.
SLI Consortium: A genomewide scan identifies two novel loci involved in specific language impairment. Am J Hum Genet. 2002, 70: 384-398. 10.1086/338649.
SLI Consortium: Highly significant linkage to the SLI1 locus in an expanded sample of individuals affected by specific language impairment. Am J Hum Genet. 2004, 74: 1225-1238.
Monaco AP: Multivariate linkage analysis of specific language impairment (SLI). Ann Hum Genet. 2007, 71: 660-673. 10.1111/j.1469-1809.2007.00361.x.
Falcaro M, Pickles A, Newbury DF, Addis L, Banfield E, Fisher SE, Monaco AP, Simkin Z, Conti-Ramsden G: Genetic and phenotypic effects of phonological short-term memory and grammatical morphology in specific language impairment. Genes Brain Behav. 2008, 7: 393-402. 10.1111/j.1601-183X.2007.00364.x.
Bartlett CW, Flax JF, Logue MW, Vieland VJ, Bassett AS, Tallal P, Brzustowicz LM: A major susceptibility locus for specific language impairment is located on 13q21. Am J Hum Genet. 2002, 71: 45-55. 10.1086/341095.
Bartlett CW, Flax JF, Logue MW, Smith BJ, Vieland VJ, Tallal P, Brzustowicz LM: Examination of potential overlap in autism and language loci on chromosomes 2, 7, and 13 in two independent samples ascertained for specific language impairment. Hum Hered. 2004, 57: 10-20. 10.1159/000077385.
Newbury DF, Winchester L, Addis L, Paracchini S, Buckingham LL, Clark A, Cohen W, Cowie H, Dworzynski K, Everitt A, Goodyer IM, Hennessy E, Kindley AD, Miller LL, Nasir J, O'Hare A, Shaw D, Simkin Z, Simonoff E, Slonims V, Watson J, Ragoussis J, Fisher SE, Seckl JR, Helms PJ, Bolton PF, Pickles A, Conti-Ramsden G, Baird G, Bishop DV, Monaco AP: CMIP and ATP2C2 modulate phonological short-term memory in language impairment. Am J Hum Genet. 2009, 85: 264-272. 10.1016/j.ajhg.2009.07.004.
Xiang M, Mohamalawari D, Rao R: A novel isoform of the secretory pathway Ca2+, Mn(2+)-ATPase, hSPCA2, has unusual properties and is expressed in the brain. J Biol Chem. 2005, 280: 11608-11614. 10.1074/jbc.M413116200.
Kamal M, Valanciute A, Dahan K, Ory V, Pawlak A, Lang P, Guellaen G, Sahali D: C-mip interacts physically with RelA and inhibits nuclear factor kappa B activity. Mol Immunol. 2009, 46: 991-998. 10.1016/j.molimm.2008.09.034.
Vernes SC, Newbury DF, Abrahams BS, Winchester L, Nicod J, Groszer M, Alarcón M, Oliver PL, Davies KE, Geschwind DH, Monaco AP, Fisher SE: A functional genetic link between distinct developmental language disorders. N Engl J Med. 2008, 359: 2337-2345. 10.1056/NEJMoa0802828.
Villanueva P, de Barbieri Z, Palomino HM, Palomino H: [High prevalence of specific language impairment in Robinson Crusoe Island. A possible founder effect]. Rev Med Chil. 2008, 136: 186-192.
Villanueva P, Newbury DF, Jara L, De Barbieri Z, Mirza G, Palomino HM, Fernandez MA, Cazier JB, Monaco AP, Palomino H: Genome-wide analysis of genetic susceptibility to language impairment in an isolated Chilean population. Eur J Hum Genet. 2011, 19: 687-695. 10.1038/ejhg.2010.251.
Fisher SE, DeFries JC: Developmental dyslexia: genetic dissection of a complex cognitive trait. Nat Rev Neurosci. 2002, 3: 767-780.
Raskind WH, Peter B, Richards T, Eckert MM, Berninger VW: The genetics of reading disabilities: from phenotypes to candidate genes. Front Psychol. 2012, 3: 601-
Graham SA, Fisher SE: Decoding the genetics of speech and language. Curr Opin Neurobiol. 2013, 23: 43-51. 10.1016/j.conb.2012.11.006.
Prasse JE, Kikano GE: Stuttering: an overview. Am Fam Physician. 2008, 77: 1271-1276.
Drayna D, Kang C: Genetic approaches to understanding the causes of stuttering. J Neurodev Disord. 2011, 3: 374-380. 10.1007/s11689-011-9090-7.
Nippold MA: Stuttering and language ability in children: questioning the connection. Am J Speech Lang Pathol. 2012, 21: 183-196. 10.1044/1058-0360(2012/11-0078).
Kang C, Drayna D: Genetics of speech and language disorders. Annu Rev Genomics Hum Genet. 2011, 12: 145-164. 10.1146/annurev-genom-090810-183119.
Raza MH, Amjad R, Riazuddin S, Drayna D: Studies in a consanguineous family reveal a novel locus for stuttering on chromosome 16q. Hum Genet. 2012, 131: 311-313. 10.1007/s00439-011-1134-2.
Raza MH, Gertz EM, Mundorff J, Lukong J, Kuster J, Schaffer AA, Drayna D: Linkage analysis of a large African family segregating stuttering suggests polygenic inheritance. Hum Genet. 2012, 132: 385-396.
Riaz N, Steinberg S, Ahmad J, Pluzhnikov A, Riazuddin S, Cox NJ, Drayna D: Genomewide significant linkage to stuttering on chromosome 12. Am J Hum Genet. 2005, 76: 647-651. 10.1086/429226.
Kang C, Riazuddin S, Mundorff J, Krasnewich D, Friedman P, Mullikin JC, Drayna D: Mutations in the lysosomal enzyme-targeting pathway and persistent stuttering. N Engl J Med. 2010, 362: 677-685. 10.1056/NEJMoa0902630.
Gilissen C, Hoischen A, Brunner HG, Veltman JA: Unlocking Mendelian disease using exome sequencing. Genome Biol. 2011, 12: 228-10.1186/gb-2011-12-9-228.
Veltman JA, Brunner HG: De novo mutations in human genetic disease. Nat Rev Genet. 2012, 13: 565-575. 10.1038/nrg3241.
Buxbaum JD, Daly MJ, Devlin B, Lehner T, Roeder K, State MW: The autism sequencing consortium: large-scale, high-throughput sequencing in autism spectrum disorders. Neuron. 2012, 76: 1052-1056. 10.1016/j.neuron.2012.12.008.
Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, Lee C, Turner EH, Smith JD, Rieder MJ, Yoshiura K, Matsumoto N, Ohta T, Niikawa N, Nickerson DA, Bamshad MJ, Shendure J: Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010, 42: 790-793. 10.1038/ng.646.
Hoischen A, van Bon BW, Rodríguez-Santiago B, Gilissen C, Vissers LE, de Vries P, Janssen I, van Lier B, Hastings R, Smithson SF, Newbury-Ecob R, Kjaergaard S, Goodship J, McGowan R, Bartholdi D, Rauch A, Peippo M, Cobben JM, Wieczorek D, Gillessen-Kaesbach G, Veltman JA, Brunner HG, de Vries BB: De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat Genet. 2011, 43: 729-731. 10.1038/ng.868.
Sirmaci A, Spiliopoulos M, Brancati F, Powell E, Duman D, Abrams A, Bademci G, Agolini E, Guo S, Konuk B, Kavaz A, Blanton S, Digilio MC, Dallapiccola B, Young J, Zuchner S, Tekin M: Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet. 2011, 89: 289-294. 10.1016/j.ajhg.2011.06.007.
Hoischen A, van Bon BW, Gilissen C, Arts P, van Lier B, Steehouwer M, de Vries P, de Reuver R, Wieskamp N, Mortier G, Devriendt K, Amorim MZ, Revencu N, Kidd A, Barbosa M, Turner A, Smith J, Oley C, Henderson A, Hayes IM, Thompson EM, Brunner HG, de Vries BB, Veltman JA: De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet. 2010, 42: 483-485. 10.1038/ng.581.
Marseglia G, Scordo MR, Pescucci C, Nannetti G, Biagini E, Scandurra V, Gerundino F, Magi A, Benelli M, Torricelli F: 372 kb Microdeletion in 18q12.3 causing SETBP1 haploinsufficiency associated with mild mental retardation and expressive speech impairment. Eur J Med Genet. 2012, 55: 216-221. 10.1016/j.ejmg.2012.01.005.
Oakley K, Han Y, Vishwakarma BA, Chu S, Bhatia R, Gudmundsson KO, Keller J, Chen X, Vasko V, Jenkins NA, Copeland NG, Du Y: Setbp1 promotes the self-renewal of murine myeloid progenitors via activation of Hoxa9 and Hoxa10. Blood. 2012, 119: 6099-6108. 10.1182/blood-2011-10-388710.
Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M, van Bon BW, Hoischen A, de Vries BB, Brunner HG, Veltman JA: A de novo paradigm for mental retardation. Nat Genet. 2010, 42: 1109-1112. 10.1038/ng.712.
O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, Rieder MJ, Nickerson DA, Bernier R, Fisher SE, Shendure J, Eichler EE: Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011, 43: 585-589. 10.1038/ng.835.
O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE: Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012, 485: 246-250. 10.1038/nature10989.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, Meyer KA, Bilguvar K, Mane SM, Sestan N, Lifton RP, Günel M, Roeder K, Geschwind DH, Devlin B, State MW: De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012, 485: 237-241. 10.1038/nature10945.
Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, Cai G, Lihm J, Dannenfelser R, Jabado O, Peralta Z, Nagaswamy U, Muzny D, Reid JG, Newsham I, Wu Y, et al: Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012, 485: 242-245. 10.1038/nature11011.
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, Ghiban E, Kramer M, Parla J, Demeter R, Fulton LL, Fulton RS, Magrini VJ, Ye K, Darnell JC, Darnell RB, Mardis ER, Wilson RK, Schatz MC, McCombie WR, Wigler M: De novo gene disruptions in children on the autistic spectrum. Neuron. 2012, 74: 285-299. 10.1016/j.neuron.2012.04.009.
Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, Ataman B, Schmitz-Abe K, Harmin DA, Adli M, Malik AN, D'Gama AM, Lim ET, Sanders SJ, Mochida GH, Partlow JN, Sunu CM, Felie JM, Rodriguez J, Nasir RH, Ware J, Joseph RM, Hill RS, Kwan BY, Al-Saffar M, Mukaddes NM, Hashmi A, Balkhy S, Gascon GG, Hisama FM, LeClair E, et al: Using whole-exome sequencing to identify inherited causes of autism. Neuron. 2013, 77: 259-273. 10.1016/j.neuron.2012.11.002.
Lim ET, Raychaudhuri S, Sanders SJ, Stevens C, Sabo A, MacArthur DG, Neale BM, Kirby A, Ruderfer DM, Fromer M, Lek M, Liu L, Flannick J, Ripke S, Nagaswamy U, Muzny D, Reid JG, Hawes A, Newsham I, Wu Y, Lewis L, Dinh H, Gross S, Wang LS, Lin CF, Valladares O, Gabriel SB, dePristo M, Altshuler DM, Purcell SM, et al: Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron. 2013, 77: 235-242. 10.1016/j.neuron.2012.12.029.
Ng PC, Henikoff S: SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31: 3812-3814. 10.1093/nar/gkg509.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR: A method and server for predicting damaging missense mutations. Nat Methods. 2010, 7: 248-249. 10.1038/nmeth0410-248.
Vernes SC, Nicod J, Elahi FM, Coventry JA, Kenny N, Coupe AM, Bird LE, Davies KE, Fisher SE: Functional genetic analysis of mutations implicated in a human speech and language disorder. Hum Mol Genet. 2006, 15: 3154-3167. 10.1093/hmg/ddl392.
Li S, Weidenfeld J, Morrisey EE: Transcriptional and DNA binding activity of the Foxp1/2/4 family is modulated by heterotypic and homotypic protein interactions. Mol Cell Biol. 2004, 24: 809-822. 10.1128/MCB.24.2.809-822.2004.
Shu W, Yang H, Zhang L, Lu MM, Morrisey EE: Characterization of a new subfamily of winged-helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. J Biol Chem. 2001, 276: 27488-27497. 10.1074/jbc.M100636200.
Spiteri E, Konopka G, Coppola G, Bomar J, Oldham M, Ou J, Vernes SC, Fisher SE, Ren B, Geschwind DH: Identification of the transcriptional targets of FOXP2, a gene linked to speech and language, in developing human brain. Am J Hum Genet. 2007, 81: 1144-1157. 10.1086/522237.
Vernes SC, Spiteri E, Nicod J, Groszer M, Taylor JM, Davies KE, Geschwind DH, Fisher SE: High-throughput analysis of promoter occupancy reveals direct neural targets of FOXP2, a gene mutated in speech and language disorders. Am J Hum Genet. 2007, 81: 1232-1250. 10.1086/522238.
Vernes SC, Oliver PL, Spiteri E, Lockstone HE, Puliyadi R, Taylor JM, Ho J, Mombereau C, Brewer A, Lowy E, Nicod J, Groszer M, Baban D, Sahgal N, Cazier JB, Ragoussis J, Davies KE, Geschwind DH, Fisher SE: Foxp2 regulates gene networks implicated in neurite outgrowth in the developing brain. PLoS Genet. 2011, 7: e1002145-10.1371/journal.pgen.1002145.
Roll P, Vernes SC, Bruneau N, Cillario J, Ponsole-Lenfant M, Massacrier A, Rudolf G, Khalife M, Hirsch E, Fisher SE, Szepetowski P: Molecular networks implicated in speech-related disorders: FOXP2 regulates the SRPX2/uPAR complex. Hum Mol Genet. 2010, 19: 4848-4860. 10.1093/hmg/ddq415.
Walker RM, Hill AE, Newman AC, Hamilton G, Torrance HS, Anderson SM, Ogawa F, Derizioti P, Nicod J, Vernes SC, Fisher SE, Thomson PA, Porteous DJ, Evans KL: The DISC1 promoter: characterization and regulation by FOXP2. Hum Mol Genet. 2012, 21: 2862-2872. 10.1093/hmg/dds111.
Mukamel Z, Konopka G, Wexler E, Osborn GE, Dong H, Bergman MY, Levitt P, Geschwind DH: Regulation of MET by FOXP2, genes implicated in higher cognitive dysfunction and autism risk. J Neurosci. 2011, 31: 11437-11442. 10.1523/JNEUROSCI.0181-11.2011.
Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, Stewart CL, Xu X, Chiu SY, Shrager P, Furley AJ, Peles E: Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003, 162: 1149-1160. 10.1083/jcb.200305018.
Anderson GR, Galfin T, Xu W, Aoto J, Malenka RC, Sudhof TC: Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc Natl Acad Sci USA. 2012, 109: 18120-18125. 10.1073/pnas.1216398109.
Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, Stephan DA, Morton DH: Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006, 354: 1370-1377. 10.1056/NEJMoa052773.
Alarcón M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, Sebat J, Wigler M, Martin CL, Ledbetter DH, Nelson SF, Cantor RM, Geschwind DH: Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008, 82: 150-159. 10.1016/j.ajhg.2007.09.005.
Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, Rea A, Guy M, Lin S, Cook EH, Chakravarti A: A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet. 2008, 82: 160-164. 10.1016/j.ajhg.2007.09.015.
Bakkaloglu B, O'Roak BJ, Louvi A, Gupta AR, Abelson JF, Morgan TM, Chawarska K, Klin A, Ercan-Sencicek AG, Stillman AA, Tanriover G, Abrahams BS, Duvall JA, Robbins EM, Geschwind DH, Biederer T, Gunel M, Lifton RP, State MW: Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am J Hum Genet. 2008, 82: 165-173. 10.1016/j.ajhg.2007.09.017.
Friedman JI, Vrijenhoek T, Markx S, Janssen IM, van der Vliet WA, Faas BH, Knoers NV, Cahn W, Kahn RS, Edelmann L, Davis KL, Silverman JM, Brunner HG, van Kessel AG, Wijmenga C, Ophoff RA, Veltman JA: CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol Psychiatry. 2008, 13: 261-266. 10.1038/sj.mp.4002049.
Stein MB, Yang BZ, Chavira DA, Hitchcock CA, Sung SC, Shipon-Blum E, Gelernter J: A common genetic variant in the neurexin superfamily member CNTNAP2 is associated with increased risk for selective mutism and social anxiety-related traits. Biol Psychiatry. 2011, 69: 825-831. 10.1016/j.biopsych.2010.11.008.
Verkerk AJ, Mathews CA, Joosse M, Eussen BH, Heutink P, Oostra BA: CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics. 2003, 82: 1-9. 10.1016/S0888-7543(03)00097-1.
Teramitsu I, Kudo LC, London SE, Geschwind DH, White SA: Parallel FoxP1 and FoxP2 expression in songbird and human brain predicts functional interaction. J Neurosci. 2004, 24: 3152-3163. 10.1523/JNEUROSCI.5589-03.2004.
Shu W, Lu MM, Zhang Y, Tucker PW, Zhou D, Morrisey EE: Foxp2 and Foxp1 cooperatively regulate lung and esophagus development. Development. 2007, 134: 1991-2000. 10.1242/dev.02846.
Leblond CS, Heinrich J, Delorme R, Proepper C, Betancur C, Huguet G, Konyukh M, Chaste P, Ey E, Rastam M, Anckarsäter H, Nygren G, Gillberg IC, Melke J, Toro R, Regnault B, Fauchereau F, Mercati O, Lemière N, Skuse D, Poot M, Holt R, Monaco AP, Järvelä I, Kantojärvi K, Vanhala R, Curran S, Collier DA, Bolton P, Chiocchetti A, et al: Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012, 8: e1002521-10.1371/journal.pgen.1002521.
Ruzzo EK, Pappas AL, Goldstein DB: Modifier genetics in neuropsychiatric disease: challenges and opportunities. Genome Biol. 2012, 13: 150-10.1186/gb-2012-13-3-150.
Vernes SC, MacDermot KD, Monaco AP, Fisher SE: Assessing the impact of FOXP1 mutations on developmental verbal dyspraxia. Eur J Hum Genet. 2009, 17: 1354-1358. 10.1038/ejhg.2009.43.
Hamdan FF, Daoud H, Rochefort D, Piton A, Gauthier J, Langlois M, Foomani G, Dobrzeniecka S, Krebs MO, Joober R, Lafrenière RG, Lacaille JC, Mottron L, Drapeau P, Beauchamp MH, Phillips MS, Fombonne E, Rouleau GA, Michaud JL: De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am J Hum Genet. 2010, 87: 671-678. 10.1016/j.ajhg.2010.09.017.
Horn D, Kapeller J, Rivera-Brugués N, Moog U, Lorenz-Depiereux B, Eck S, Hempel M, Wagenstaller J, Gawthrope A, Monaco AP, Bonin M, Riess O, Wohlleber E, Illig T, Bezzina CR, Franke A, Spranger S, Villavicencio-Lorini P, Seifert W, Rosenfeld J, Klopocki E, Rappold GA, Strom TM: Identification of FOXP1 deletions in three unrelated patients with mental retardation and significant speech and language deficits. Hum Mutat. 2010, 31: E1851-1860. 10.1002/humu.21362.
Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A, Ernst C, Hanscom C, Rossin E, Lindgren AM, Pereira S, Ruderfer D, Kirby A, Ripke S, Harris DJ, Lee JH, Ha K, Kim HG, Solomon BD, Gropman AL, Lucente D, Sims K, Ohsumi TK, Borowsky ML, Loranger S, Quade B, Lage K, Miles J, Wu BL, Shen Y, et al: Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012, 149: 525-537. 10.1016/j.cell.2012.03.028.
Lee WS, Kang C, Drayna D, Kornfeld S: Analysis of mannose 6-phosphate uncovering enzyme mutations associated with persistent stuttering. J Biol Chem. 2011, 286: 39786-39793. 10.1074/jbc.M111.295899.
Cooper GM, Shendure J: Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat Rev Genet. 2011, 12: 628-640. 10.1038/nrg3046.
Fowler DM, Araya CL, Fleishman SJ, Kellogg EH, Stephany JJ, Baker D, Fields S: High resolution mapping of protein sequence-function relationships. Nat Methods. 2010, 7: 741-746. 10.1038/nmeth.1492.
Patwardhan RP, Lee C, Litvin O, Young DL, Pe'er D, Shendure J: High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat Biotech. 2009, 27: 1173-1175. 10.1038/nbt.1589.
CAS Google Scholar
Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, Smith RP, May D, Lee C, Andrie JM, Lee SI, Cooper GM, Ahituv N, Pennacchio LA, Shendure J: Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 2012, 30: 265-270. 10.1038/nbt.2136.
Pitt JN, Ferré-D'Amaré AR: Rapid construction of empirical RNA fitness landscapes. Science. 2010, 330: 376-379. 10.1126/science.1192001.
ENCODE Project Consortium, Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, Khatun J, Lajoie BR, Landt SG, Lee BK, Pauli F, Rosenbloom KR, Sabo P, Safi A, Sanyal A, Shoresh N, Simon JM, Song L, Trinklein ND, Altshuler RC, Birney E, Brown JB, Cheng C, Djebali S, Dong X, et al: An integrated encyclopedia of DNA elements in the human genome. Nature. 2012, 489: 57-74. 10.1038/nature11247.
Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, Cherry JM, Snyder M: Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22: 1790-1797. 10.1101/gr.137323.112.
White SA, Fisher SE, Geschwind DH, Scharff C, Holy TE: Singing mice, songbirds, and more: models for FOXP2 function and dysfunction in human speech and language. J Neurosci. 2006, 26: 10376-10379. 10.1523/JNEUROSCI.3379-06.2006.
Lai CS, Gerrelli D, Monaco AP, Fisher SE, Copp AJ: FOXP2 expression during brain development coincides with adult sites of pathology in a severe speech and language disorder. Brain. 2003, 126: 2455-2462. 10.1093/brain/awg247.
Belton E, Salmond CH, Watkins KE, Vargha-Khadem F, Gadian DG: Bilateral brain abnormalities associated with dominantly inherited verbal and orofacial dyspraxia. Hum Brain Mapp. 2003, 18: 194-200. 10.1002/hbm.10093.
Liegeois F, Baldeweg T, Connelly A, Gadian DG, Mishkin M, Vargha-Khadem F: Language fMRI abnormalities associated with FOXP2 gene mutation. Nat Neurosci. 2003, 6: 1230-1237. 10.1038/nn1138.
Ferland RJ, Cherry TJ, Preware PO, Morrisey EE, Walsh CA: Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J Comp Neurol. 2003, 460: 266-279. 10.1002/cne.10654.
Enard W: FOXP2 and the role of cortico-basal ganglia circuits in speech and language evolution. Curr Opin Neurobiol. 2011, 21: 415-424. 10.1016/j.conb.2011.04.008.
Groszer M, Keays DA, Deacon RM, de Bono JP, Prasad-Mulcare S, Gaub S, Baum MG, French CA, Nicod J, Coventry JA, Enard W, Fray M, Brown SD, Nolan PM, Pääbo S, Channon KM, Costa RM, Eilers J, Ehret G, Rawlins JN, Fisher SE: Impaired synaptic plasticity and motor learning in mice with a point mutation implicated in human speech deficits. Curr Biol. 2008, 18: 354-362. 10.1016/j.cub.2008.01.060.
French CA, Jin X, Campbell TG, Gerfen E, Groszer M, Fisher SE, Costa RM: An aetiological Foxp2 mutation causes aberrant striatal activity and alters plasticity during skill learning. Mol Psychiatry. 2011, 17: 1077-1085.
Kurt S, Groszer M, Fisher SE, Ehret G: Modified sound-evoked brainstem potentials in Foxp2 mutant mice. Brain Res. 2009, 1289: 30-36.
Kurt S, Fisher SE, Ehret G: Foxp2 mutations impair auditory-motor association learning. PLoS One. 2012, 7: e33130-10.1371/journal.pone.0033130.
Watkins KE, Dronkers NF, Vargha-Khadem F: Behavioural analysis of an inherited speech and language disorder: comparison with acquired aphasia. Brain. 2002, 125: 452-464. 10.1093/brain/awf058.
Scharff C, Adam I: Neurogenetics of birdsong. Curr Opin Neurobiol. 2013, 23: 29-36. 10.1016/j.conb.2012.10.001.
Haesler S, Wada K, Nshdejan A, Morrisey EE, Lints T, Jarvis ED, Scharff C: FoxP2 expression in avian vocal learners and non-learners. J Neurosci. 2004, 24: 3164-3175. 10.1523/JNEUROSCI.4369-03.2004.
Miller JE, Spiteri E, Condro MC, Dosumu-Johnson RT, Geschwind DH, White SA: Birdsong decreases protein levels of FoxP2, a molecule required for human speech. J Neurophysiol. 2008, 100: 2015-2025. 10.1152/jn.90415.2008.
Teramitsu I, Poopatanapong A, Torrisi S, White SA: Striatal FoxP2 is actively regulated during songbird sensorimotor learning. PLoS One. 2010, 5: e8548-10.1371/journal.pone.0008548.
Teramitsu I, White SA: FoxP2 regulation during undirected singing in adult songbirds. J Neurosci. 2006, 26: 7390-7394. 10.1523/JNEUROSCI.1662-06.2006.
Haesler S, Rochefort C, Georgi B, Licznerski P, Osten P, Scharff C: Incomplete and inaccurate vocal imitation after knockdown of FoxP2 in songbird basal ganglia nucleus Area X. PLoS Biol. 2007, 5: e321-10.1371/journal.pbio.0050321.
Schulz SB, Haesler S, Scharff C, Rochefort C: Knockdown of FoxP2 alters spine density in Area X of the zebra finch. Genes Brain Behav. 2010, 9: 732-740. 10.1111/j.1601-183X.2010.00607.x.
Zweier C, de Jong EK, Zweier M, Orrico A, Ousager LB, Collins AL, Bijlsma EK, Oortveld MA, Ekici AB, Reis A, Schenck A, Rauch A: CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet. 2009, 85: 655-666. 10.1016/j.ajhg.2009.10.004.
Gregor A, Albrecht B, Bader I, Bijlsma EK, Ekici AB, Engels H, Hackmann K, Horn D, Hoyer J, Klapecki J, Kohlhase J, Maystadt I, Nagl S, Prott E, Tinschert S, Ullmann R, Wohlleber E, Woods G, Reis A, Rauch A, Zweier C: Expanding the clinical spectrum associated with defects in CNTNAP2 and NRXN1. BMC Med Genet. 2011, 12: 106-10.1186/1471-2350-12-106.
Newbury DF, Paracchini S, Scerri TS, Winchester L, Addis L, Richardson AJ, Walter J, Stein JF, Talcott JB, Monaco AP: Investigation of dyslexia and SLI risk variants in reading- and language-impaired subjects. Behav Genet. 2011, 41: 90-104. 10.1007/s10519-010-9424-3.
Whitehouse AJ, Bishop DV, Ang QW, Pennell CE, Fisher SE: CNTNAP2 variants affect early language development in the general population. Genes Brain Behav. 2011, 10: 451-456. 10.1111/j.1601-183X.2011.00684.x.
Whalley HC, O'Connell G, Sussmann JE, Peel A, Stanfield AC, Hayiou-Thomas ME, Johnstone EC, Lawrie SM, McIntosh AM, Hall J: Genetic variation in CNTNAP2 alters brain function during linguistic processing in healthy individuals. Am J Med Genet B Neuropsychiatr Genet. 2011, 156B: 941-948.
Kos M, van den Brink D, Snijders TM, Rijpkema M, Franke B, Fernandez G, Hagoort P: CNTNAP2 and language processing in healthy individuals as measured with ERPs. PLoS One. 2012, 7: e46995-10.1371/journal.pone.0046995.
Dennis EL, Jahanshad N, Rudie JD, Brown JA, Johnson K, McMahon KL, de Zubicaray GI, Montgomery G, Martin NG, Wright MJ, Bookheimer SY, Dapretto M, Toga AW, Thompson PM: Altered structural brain connectivity in healthy carriers of the autism risk gene, CNTNAP2. Brain Connect. 2011, 1: 447-459. 10.1089/brain.2011.0064.
Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, Hansen NF, Durand EY, Malaspinas AS, Jensen JD, Marques-Bonet T, Alkan C, Prüfer K, Meyer M, Burbano HA, Good JM, Schultz R, Aximu-Petri A, Butthof A, Höber B, Höffner B, Siegemund M, Weihmann A, Nusbaum C, Lander ES, Russ C, et al: A draft sequence of the Neandertal genome. Science. 2010, 328: 710-722. 10.1126/science.1188021.
Meyer M, Kircher M, Gansauge MT, Li H, Racimo F, Mallick S, Schraiber JG, Jay F, Prüfer K, de Filippo C, Sudmant PH, Alkan C, Fu Q, Do R, Rohland N, Tandon A, Siebauer M, Green RE, Bryc K, Briggs AW, Stenzel U, Dabney J, Shendure J, Kitzman J, Hammer MF, Shunkov MV, Derevianko AP, Patterson N, Andrés AM, Eichler EE, et al: A high-coverage genome sequence from an archaic Denisovan individual. Science. 2012, 338: 222-226. 10.1126/science.1224344.
Enard W, Przeworski M, Fisher SE, Lai CS, Wiebe V, Kitano T, Monaco AP, Paabo S: Molecular evolution of FOXP2, a gene involved in speech and language. Nature. 2002, 418: 869-872. 10.1038/nature01025.
Maricic T, Günther V, Georgiev O, Gehre S, Curlin M, Schreiweis C, Naumann R, Burbano HA, Meyer M, Lalueza-Fox C, de la Rasilla M, Rosas A, Gajovic S, Kelso J, Enard W, Schaffner W, Pääbo S: A recent evolutionary change affects a regulatory element in the human FOXP2 gene. Mol Biol Evol. 2012, 30: 844-852.
Falivelli G, De Jaco A, Favaloro FL, Kim H, Wilson J, Dubi N, Ellisman MH, Abrahams BS, Taylor P, Comoletti D: Inherited genetic variants in autism-related CNTNAP2 show perturbed trafficking and ATF6 activation. Hum Mol Genet. 2012, 21: 4761-4773. 10.1093/hmg/dds320.
Download references
Author information
Authors and affiliations.
Max Planck Institute for Psycholinguistics, 6525 XD, Nijmegen, The Netherlands
Pelagia Deriziotis & Simon E Fisher
Donders Institute for Brain, Cognition and Behaviour, Radboud University, 6525 EN, Nijmegen, The Netherlands
Simon E Fisher
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Simon E Fisher .
Additional information
Competing interests.
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Authors’ original file for figure 1
Rights and permissions.
Reprints and permissions
About this article
Cite this article.
Deriziotis, P., Fisher, S.E. Neurogenomics of speech and language disorders: the road ahead. Genome Biol 14 , 204 (2013). https://doi.org/10.1186/gb-2013-14-4-204
Download citation
Published : 18 April 2013
DOI : https://doi.org/10.1186/gb-2013-14-4-204
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- functional validation
- neurodevelopmental disorders
Genome Biology
ISSN: 1474-760X
- Submission enquiries: [email protected]
- General enquiries: [email protected]
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Published: 27 April 2021
Speech and language deficits are central to SETBP1 haploinsufficiency disorder
- Angela Morgan ORCID: orcid.org/0000-0003-1147-7405 1 , 2 , 3 , 4 , 5 na1 ,
- Ruth Braden 1 , 2 , 5 na1 ,
- Maggie M. K. Wong ORCID: orcid.org/0000-0002-9438-0141 6 ,
- Estelle Colin ORCID: orcid.org/0000-0001-7913-3938 7 ,
- David Amor 1 , 3 , 4 ,
- Frederique Liégeois 8 ,
- Siddharth Srivastava ORCID: orcid.org/0000-0001-7008-1879 9 ,
- Adam Vogel 10 ,
- Varoona Bizaoui 11 ,
- Kara Ranguin ORCID: orcid.org/0000-0002-9732-8090 11 ,
- Simon E. Fisher ORCID: orcid.org/0000-0002-3132-1996 6 , 12 &
- Bregje W. van Bon 13
European Journal of Human Genetics volume 29 , pages 1216–1225 ( 2021 ) Cite this article
1826 Accesses
21 Citations
30 Altmetric
Metrics details
- Behavioural genetics
- Development
- Genetics research
Expressive communication impairment is associated with haploinsufficiency of SETBP1 , as reported in small case series. Heterozygous pathogenic loss-of-function (LoF) variants in SETBP1 have also been identified in independent cohorts ascertained for childhood apraxia of speech (CAS), warranting further investigation of the roles of this gene in speech development. Thirty-one participants (12 males, aged 0; 8–23; 2 years, 28 with pathogenic SETBP1 LoF variants, 3 with 18q12.3 deletions) were assessed for speech, language and literacy abilities. Broader development was examined with standardised motor, social and daily life skills assessments. Gross and fine motor deficits (94%) and intellectual impairments (68%) were common. Protracted and aberrant speech development was consistently seen, regardless of motor or intellectual ability. We expand the linguistic phenotype associated with SETBP1 LoF syndrome (SETBP1 haploinsufficiency disorder), revealing a striking speech presentation that implicates both motor (CAS, dysarthria) and language (phonological errors) systems, with CAS (80%) being the most common diagnosis. In contrast to past reports, the understanding of language was rarely better preserved than language expression (29%). Language was typically low, to moderately impaired, with commensurate expression and comprehension ability. Children were sociable with a strong desire to communicate. Minimally verbal children (32%) augmented speech with sign language, gestures or digital devices. Overall, relative to general development, spoken language and literacy were poorer than social, daily living, motor and adaptive behaviour skills. Our findings show that poor communication is a central feature of SETBP1 haploinsufficiency disorder, confirming this gene as a strong candidate for speech and language disorders.
You have full access to this article via your institution.
Similar content being viewed by others

CDK13-related disorder: a deep characterization of speech and language abilities and addition of 33 novel cases
Lottie D. Morison, Olivia van Reyk, … Angela T. Morgan

Speech and language phenotype in Phelan-McDermid (22q13.3) syndrome
Amanda Brignell, Conway Gu, … Angela T. Morgan

Expanding the speech and language phenotype in Koolen-de Vries syndrome: late onset and periodic stuttering a novel feature
Miya St John, Olivia van Reyk, … Angela T. Morgan
Introduction
Clinical investigations of individuals with disruptions of SET binding protein 1 ( SETBP1 ) on 18q12.3 have suggested the gene as a candidate for expressive speech disorder [ 1 ]. Haploinsufficiency of SETBP1 was first associated with expressive language ‘delay’ or ‘impairment’ in descriptive single cases [ 1 , 2 ] and case series [ 3 ]. Other presenting features were mild-to-severely impaired intellect, gross and fine motor delays and/or deficits, hypotonia, distinctive facial features, attention deficits, and less commonly, autistic traits [ 1 , 3 ].
A novel disorder encompassing this symptomatology was later confirmed in a cohort selected for intellectual disability (ID) [ 4 ]. Five individuals with LoF variants and one with a de novo deletion encompassing SETBP1 were identified [ 4 ]. The authors combined these cases with two further novel cases with truncating variants from a separate ID screen, together with the previously published small deletions [ 1 , 3 ] and de novo variants [ 2 ] to examine the phenotype across all the identified individuals. Only retrospective clinical data were available, limiting the scope of the investigation. Most of the individuals were reported to have intelligence quotient and language deficits, with completely absent or significantly impaired speech in 92% of this group [ 4 ]. The nature of the speech deficits was not described, although apraxia was noted previously in one of the cases [ 1 ]. The specific speech and language phenotype in individuals with SETBP1 LoF variants remains unclear.
From a clinical genetics perspective, there is a need to identify genes that contribute to severe and persistent communication deficits, such as childhood apraxia of speech (CAS). Parental concern for speech development is a common reason for referral to paediatricians, yet the aetiology and prognosis for CAS are poorly understood and children are largely managed with a ‘watch-and-wait’ approach [ 5 ]. Until recently, few additional candidates for CAS had been revealed, since the unearthing of FOXP2 almost 20 years ago [ 6 ]. A number of new contenders have now been identified through next-generation sequencing screens of genomes/exomes in two cohorts ascertained on the basis of CAS [ 7 , 8 ]. Notably, whilst modest in cohort size ( n = 18 and n = 34, respectively), each study independently identified an individual with a heterozygous pathogenic SETBP1 LoF variant, suggesting disruptions of this gene as a recurrent cause for CAS; which occurs at a rate of only 1 or 2 cases per 1000 in the general population [ 9 ]. The findings add weight to the premise that SETBP1 may play an important role in speech and language development.
Two studies examining common single-nucleotide polymorphisms (SNPs) further support the potential relevance of SETBP1 variation for communication abilities. Associations between SETBP1 and scores on a test examining syntactic complexity (mean length of sentences and use of complex sentence structures) were reported in a Genome Wide Association Study of developmental language disorder in a geographically isolated Russian cohort aged 3–18 years [ 10 ]. More tentatively, SNPs in SETBP1 have been reported to show association with phonological working memory; just one of many reading-related traits examined across a reading-impaired cohort of modest size for complex trait analyses ( n = 135) [ 11 ]. Whether SETBP1 is more closely linked with variations in speech, language and/or reading ability in the general population is not yet clear.
Thus, nascent evidence from a range of sources suggests SETBP1 as a gene of relevance to speech and language development. Yet information concerning the clinical phenotype associated with SETBP1 haploinsufficiency has so far been drawn only from descriptive case series, relying largely on retrospective examination of medical records. Here, we performed in-depth examination of speech, language and literacy abilities in a cohort of 31 individuals with SETBP1 LoF variants and deletions, using standardised tests, to precisely characterise the communication phenotype of this syndrome. Linguistic performance was considered relative to other areas of neurodevelopment (e.g. motor abilities, social skills), to determine whether communication was differentially affected.
Inclusion criteria were a molecular diagnosis of heterozygous SETBP1 truncating (stop-gain or frameshift) variants or 18q12.3 deletions in individuals aged ≥6 months. Participants were recruited globally via the SETBP1 Society ( http://www.setbp1.org ) and clinician referral. Ethics approval was obtained from the Royal Children’s Hospital, Melbourne, Human Research Ethics Committee (HREC 37353A).
Thirty-one participants were recruited (Table 1 ; 20 males, average age 9 years, range 0; 8–23; 2 years). Genotypes included single-nucleotide variants (28 participants), a 18q12.3 intragenic deletion and two larger deletions encompassing SETBP1 (Fig. 1 ). Twenty four participants were novel cases, not previously reported in the literature. Nineteen participants also participated in a separate study on the broader medical phenotype (see the companion paper by Jansen et al., Eur J Hum Genet, submitted). Deletions and phenotypic data were submitted to Decipher ( https://decipher.sanger.ac.uk/ ) and sequence variants were submitted to Leiden Open Variation Database (Database ID: #chr18_002464-002468, #SETBP1_000018-000020, #SETBP1_000033, #SETBP1_000078, #SETBP1_000083, #SETBP1_000085, #SETBP1_000103, #SETBP1_000106, #SETBP1_000108-000111, #SETBP1_00014, #SETBP1_000116-000117, #SETBP1_000119-000120, #SETBP1_000123-000125, #SETBP1_000127, #SETBP1_000129).

Schematic representation of the SETBP1 protein (UniProt: Q9Y6X0) indicating loss-of-function variants included in this study. Five exons (black bars) encode isoform A of the protein (1596 amino acids). Five exons (black bars) encode isoform A of the protein (1596 amino acid residues). The SETBP1 protein sequence contains three AT-hook domains (Ath; orange; amino acids 584–596, 1016–1028, 1451–1463), a SKI homologous region (SKI; green; amino acids 706–917), a HCF1-binding motif (HCF; magenta; amino acids 991–994), a SET-binding domain (SET; blue; amino acids 1292–1488), three bipartite NLS motifs (black; amino acids 462–477, 1370–1384, 1383–1399), six PEST sequences (brown; amino acids 1–13, 269–280, 548–561, 678–689, 806–830, 1502–1526) and a repeat domain (Rpt; grey; amino acids 1520–1543) [31–34]. Blue circles represent previously reported variants and yellow circles indicate novel variants. Two individuals with larger deletions (IND 4, 24) are not shown here. For cDNA annotation of the variants see Table 1.
Health and development
Health and medical information, including data on neurodevelopmental conditions, intellectual ability and intervention (e.g. speech therapy, physiotherapy), was collected via an established survey [ 12 , 13 ] (Table 1 ), translated into multiple languages. Health professional reports (e.g. psychology, neurology, clinical genetic, speech pathology) and telehealth consults confirmed questionnaire responses. Feeding (Child Oral and Motor Proficiency Scale) [ 14 ] and drooling (Drooling Impact Scale) [ 15 ] measures were collected where age appropriate.
Verbal children completed the Diagnostic Evaluation of Articulation and Phonology to examine articulation and phonological errors [ 16 ]. A 5-minute speech sample was analysed for diagnoses of CAS [ 17 ] and dysarthria using established methods [ 18 , 19 ]. The Intelligibility in Context Scale [ 20 ] examined how often the individual is understood, with a 5 point scale of responses ranging from never to always.
Verbal children were assessed with the Children’s Communication Checklist (CCC-2) [ 21 ]. Minimally verbal children (defined as <50 spoken words), and those <4 years of age were assessed with the Macarthur Bates Communicative Development Inventory (MB-CDI) [ 22 ] and Communication and Symbolic Behaviour Scales Developmental Profile (CSBS-DP) [ 23 ]. The MB-CDI measures understanding and use of gesture, vocabulary and sentences. The CSBS-DP provides social communication, speech and symbolic communication scores for children aged 6–24 months, or for chronologically older children with limited linguistic abilities [ 23 ].
Adaptive behaviour
The Vineland Adaptive Behaviour Scales-Parent/Caregiver [ 24 ], provided domain scores for communication, socialisation, daily living and motor skills, with an overall adaptive behaviour composite. Wilcoxon signed-rank sum tests were used to determine the relative involvement of language compared to other Vineland subdomain scores.
The cohort consisted of one infant, seven pre-school children, 21 school-aged children and adolescents and two adults ( n = 31 (20 males, 11 females); Table 1 ). Participants were recruited from the US ( n = 12), Netherlands ( n = 8), United Kingdom ( n = 3), France ( n = 3), Canada ( n = 2), Israel ( n = 2) and Australia ( n = 1). Developmental issues relevant to speech and language development included early feeding difficulties (58%), and excessive drooling (35%). Almost all participants (94%) had generalised motor delay or disorder that required occupational therapy and/or physiotherapy (87%; Table 1 and Fig. 2 ). Motor deficits included difficulties with personal care (managing buttons and zippers, teeth brushing, washing), writing, drawing, using scissors, riding a bike and toilet training. Intellectual impairment was reported in the majority of individuals aged >4 years. In the seven individuals aged <4 years, six parents reported that their child was experiencing developmental delay compared to same-aged peers. Seven patients had seizures; four had febrile seizures, one had generalised tonic-clonic seizures, one had absence seizures and one reported a history of seizures despite a normal EEG. Hearing impairment was infrequent (3/31, 10%) and all presentations were mild (25–39 dBHL) and bilateral, with two cases of mixed (IND 8, 12) and one of conductive (IND 27) hearing loss, although periodic conductive losses due to otitis media were also common (58%). Visual impairments (42%) were addressed with glasses and hypermetropia was the most common diagnosis (62%). Palatal abnormalities included cleft lip and palate (IND 26), submucous cleft palate (IND 23) and a high arch palate (IND 31). Micrognathia (3/31, 10%) was noted in few participants.

2 children <2 years and hence too young for reliably determining the presence of these comorbid features.
Attention issues were common (55%; including nine with formal ADHD diagnoses, see Fig. 2 ). All but the 8-month old participant had been directly assessed for autism spectrum disorder (ASD), but only three (10%) received a clinical ASD diagnosis. Other diagnoses included developmental coordination disorder (19%) and sensory processing disorder (23%).
Academically, eight of the 21 (38%) school-aged participants attended mainstream schools and the remaining 13 (62%) attended special education schools. Of the pre-school participants, three attended mainstream, and four specialised, childcare or pre-school settings, and one was cared for at home. Learning support was common (86%) across all settings. The two young adults had completed school and were engaged in supported employment. Most parents of school-aged children and young adults (21/23, 91%) reported that their child’s academic progress had been limited by their speech and language difficulties.
Speech development was characterised by limited babbling and a reduced phonetic (sound) inventory relative to peers across the first 7 years of life; when a full inventory is typically acquired. Most participants had acquired first spoken words by 18 months of age (52%) (Table 2 ). For the majority of the verbal children, short phrases or sentences were developed by 6–7 years (protracted relative to typical developmental milestone of 2–3 years) (Table 2 ). Most (94%) had accessed speech therapy, with the exception of two young participants (aged 8 months and 1 year 3 months). The dose of speech therapy increased during the pre-school period; typically once per week/fortnight, but up to five times per week where available. Intelligibility across the group ranged from never understood (32%), to rarely (23%), sometimes (61%) and usually understood (13%), based on the Intelligibility in Context Scale [ 20 ] scores.
Verbal children presented with a complex motor speech disorder, best characterised as CAS (80%; Table 2 ) with: inconsistency of phoneme production; increased errors with increasing word length; simplified syllable structures relative to age, as well as vowel and prosodic errors. A small proportion (16%) had dysarthria, typically characterised here by low pitch, hypernasality, monotonous, monoloud and flaccid, slow speech. Other speech diagnoses of phonological disorder (48%), articulation impairment (specifically a lisp) (9%) and stuttering (3%) were reported alongside CAS.
Minimally verbal children (11/31, 35%; Table 2 ) had few spoken words but had communicative intent, and used gesture, sign and/or communication devices for expression. Of this group, six were young children (aged 3–5 years of age) and five were older (aged 7–15 years). Speech intervention for the younger group focused on language stimulation, non-verbal gestures and verbal speech production. The older group was producing single words or short phrases using their digital devices. Relative strengths in social and symbolic language abilities (average standard scores 9.29 and 9.57, where mean = 10, standard deviation = 3) relative to speech (average composite standard score 6.57) were revealed on the CSBS.
Language (expressive, receptive, written, social)
For most participants, expressive and receptive language abilities were commensurate with each other (18/28; 64%) on the VABS. Poorer expressive than receptive performance was the next most common profile (8/28; 29%). For children of reading age, written language ranged from typical (2/25; 8%) to moderately low (8/25; 32%) and low to severe (15/25; 60%). Many had difficulty with writing tasks, such as copying letters or their name, although a few older patients were able to write in longer sentences. A large proportion (10/23; 43%) had received a formal diagnosis of a reading and/or writing disorder from a health professional.
The CCC-2 enabled further comparison within and across general communication (e.g. semantics, syntax, coherence) and social interaction domains. All children assessed with the CCC-2 ( n = 16), had poor communication abilities (Table 3 ). Pragmatic language and social skills were relative strengths overall, compared to speech, language structure, vocabulary and discourse, based on individual scaled scores (Fig. 3 ). Autistic traits were reported in half this group (8/16, 50%), including poor social skills and restricted interests compared to peers. Yet only three had a clinical diagnosis of ASD, as noted earlier. Participants showed a desire to communicate and share interests, with intact basic social skills and non-verbal gestures. Whilst data are limited, a widening gap in social skills was suggested, relative to peers, with increasing age (Table 2 ).

Lines denote median scores and X denotes the mean scores; • indicates an outlier. Scaled scores between 7 and 13 are within the average range.
Adaptive behaviour: language relative to daily functioning, social and motor skills
Overall adaptive behaviour scores were in the moderately low range (Table 4 ). This was commensurate with daily living skills and socialisation (Table 4 and Fig. 4 ). Motor skills were stronger than communication abilities ( p = 0.0021), although these data represent participants aged <9; 11 only, with information about normative motor skills unavailable for older children. Fine motor skills were poorer relative to gross motor in most cases, confirming parent reported motor abilities from the questionnaire data. Performance in the communication domain was substantially lower than that for socialisation ( p = 0.0055) and daily living skills ( p = 0.0023) domains (Table 4 ).

Lines denote median scores; X denotes mean scores; • indicates an outlier; ABC adaptive behaviour composite, that is overall combined score. Standard scores between 85 and 115 are considered within the average range.
Here we report the speech and language phenotype of individuals selected for pathogenic SETBP1 LoF variants. Our findings indicate that articulatory, spoken and written language (reading, writing) deficits are distinctive features of the broader neurodevelopmental profile. We expand the phenotype of this disorder beyond ‘expressive speech’ difficulties, to reveal specific sub-types of speech disorder and highlight difficulties with the understanding as well as expression of language.
Protracted and impoverished speech, language and literacy (reading, writing) development was seen across the group, regardless of cognitive ability. Although first words were developed at the typical 12-month milestone for some, the ongoing trajectory of linguistic development was markedly protracted. Verbal children displayed a complex range of speech diagnoses implicating perturbed motor (CAS, articulation impairment, dysarthria) and linguistic deficits (phonological errors) that have not previously been recognized as features of SETBP1 haploinsufficiency disorder. Yet CAS was the most common finding in our cohort, in line with the recent identification of pathogenic SETBP1 LoF variants in gene discovery cohorts ascertained for CAS [ 7 , 8 ]. The co-morbid articulation, phonological and dysarthric impairments seen alongside CAS were more notable in older children in the middle school years. Changes in speech profile across the lifespan are recognised in other neurogenetic conditions [ 8 , 12 , 25 ] and confirm the need for regular speech surveillance to enable precisely targeted therapies at particular ages.
A subset of children remained minimally verbal at ages 7–15 years. How to extricate the relative cognitive-linguistic from motor contributions in children with minimally verbal presentations is an area of ongoing debate in other neurodevelopmental conditions such as ASD [ 26 ], and no simple algorithm is available. All minimally verbal children here had communicative intent and used augmentative approaches alongside speech, such as sign language, gesture or digital devices to convey messages. This strong desire to communicate was also reported in the histories of children who became verbal, showing little differentiation between verbal and minimally verbal speakers in this regard. One hypothesis to explain the minimally verbal presentation in some is that they may have more severe involvement on the speech-motor continuum, described as anarthria and/or significant speech praxis. Early speech intervention appears to be critical for all with SETBP1 LoF variants who present with severe speech disorder, with best evidenced approaches for speech apraxia known to involve intensive therapy as often as four sessions per week [ 27 ]. Future clinical trials of intensive speech therapies in individuals with SETBP1 LoF variants are warranted.
In terms of language performance; previous case descriptions of children with SETBP1 LoF variants have implied that language comprehension is more intact than language production [ 1 , 3 ]. Yet administration of standardised language tests in our cohort revealed that understanding of language is largely commensurate with expression. This highlights potential for clinical bias in making subjective assessments of language comprehension in children with speech production disorders. Further, language deficits appeared ubiquitous without clear disparity across sub-domains of vocabulary, syntax and coherence. Similarly, there was corresponding involvement of written (reading, spelling) and spoken language, without clear dissociations between these skills. Spoken and written language abilities were in turn, generally commensurate with cognitive abilities.
In terms of broader neurodevelopmental profile, clinical reports of gross and fine motor deficits affecting motor planning, programming and execution occurred with equivalent frequency to the speech-motor deficits seen here. Attention deficits and cognitive impairment were also prevalent. These are recognisable features previously reported as concomitant with CAS [ 8 ].
Differentiating severe communication deficits from ASD can be challenging [ 28 ] and for some individuals, the negative cycle of communication breakdown leads to further social withdrawal over time [ 29 , 30 ]. A number of children in our cohort had ‘autistic features’ represented by limited social skills and restricted interests relative to peers, yet only three had a formal ASD diagnosis. Further, concern over limited speech development, rather than autistic features, was the core presenting concern for parents, and all had a strong desire to communicate, despite their recognised social skill deficits. Overall, we found limited evidence for a distinct ASD signature associated with SETBP1 LoF variants.
Clinical implications
We show that aberrant communication development is a central feature of the SETBP1 LoF syndrome. Children with heterozygous pathogenic SETBP1 LoF variants or deletions should be enrolled in speech therapy in the first year of life. Given the markedly delayed verbal communication trajectory, multi-modal communication, such as sign language or communication devices would support language acquisition prior to speech developing. The complex and widespread linguistic deficits signal that children will need speech-motor therapies to develop verbal speech, but also phonological interventions focused on early literacy awareness and approaches targeting language comprehension as well as production. Whilst children demonstrate a strong desire to communicate, social skills warrant therapeutic attention. Given this pervasive communication profile, we confirm SETBP1 as a strong candidate for speech and language disorders.
Marseglia G, Scordo MR, Pescucci C, Nannetti G, Biagini E, Scandurra V, et al. 372 kb microdeletion in 18q12.3 causing SETBP1 haploinsufficiency associated with mild mental retardation and expressive speech impairment. Eur J Med Genet. 2012;55:216–21.
Article Google Scholar
Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–82.
Article CAS Google Scholar
Filges I, Shimojima K, Okamoto N, Röthlisberger B, Weber P, Huber AR, et al. Reduced expression by SETBP1 haploinsufficiency causes developmental and expressive language delay indicating a phenotype distinct from Schinzel-Giedion syndrome. J Med Genet. 2011;48:117–22.
Coe BP, Witherspoon K, Rosenfeld JA, van Bon BWM, Vulto-van Silfhout AT, Bosco P, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet. 2014;46:1063–71.
Morgan AT, Eecen KT, Pezic A, Brommeyer K, Mei C, Eadie P, et al. Who to refer for speech therapy at 4 years of age versus who to “watch and wait”? J Pediatr. 2017;185:200–4.
Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature 2001;413:519–23.
Eising E, Carrion-Castillo A, Vino A, Strand EA, Jakielski KJ, Scerri TS, et al. A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol Psychiatry. 2018;24:1065–78.
Hildebrand MS, Jackson VE, Scerri TS, Van Reyk O, Coleman M, Braden RO, et al. Severe childhood speech disorder: gene discovery highlights transcriptional dysregulation. Neurology. 2020;94:e2148–67. https://doi.org/10.1212/WNL.0000000000009441 .
Article PubMed Google Scholar
Shriberg LD. Five subtypes of developmental phonological disorders. Clin Commun Disord. 1994;4:38–53.
CAS PubMed Google Scholar
Kornilov SA, Rakhlin N, Koposov R, Lee M, Yrigollen C, Caglayan AO, et al. Genome-wide association and exome sequencing study of language disorder in an isolated population. Pediatrics. 2016;137:e20152469. https://doi.org/10.1542/peds.2015-2469 .
Article PubMed PubMed Central Google Scholar
Perdue MV, Mascheretti S, Kornilov SA, Jasińska KK, Ryherd K, Mencl WE, et al. Common variation within the SETBP1 gene is associated with reading-related skills and patterns of functional neural activation. Neuropsychologia 2019;130:44–51.
Morgan AT, van Haaften L, van Hulst K, Edley C, Mei C, Tan TY, et al. Early speech development in Koolen de Vries syndrome limited by oral praxis and hypotonia. Eur J Hum Genet. 2018;26:75–84.
Mei C, Fedorenko E, Amor DJ, Boys A, Hoeflin C, Carew P, et al. Deep phenotyping of speech and language skills in individuals with 16p11.2 deletion. Eur J Hum Genet. 2018;26:676–86.
Pados BF, Thoyre SM, Park J. Age-based norm-reference values for the Child Oral and Motor Proficiency Scale. Acta Paediatrica. 2018;107:1427–32.
Reid SM, Johnson HM, Reddihough DS. The Drooling Impact Scale: a measure of the impact of drooling in children with developmental disabilities. Dev Med Child Neurol. 2010;52:e23–8.
Dodd B, Hua Z, Crosbie S, Holm A, Ozanne A. Diagnostic evaluation of articulation and phonology. London: Psychological Corporation; 2002.
American-Speech-Language-Hearing-Association. Childhood apraxia of speech. 2007. http://www.asha.org .
Duffy JR. Motor speech disorders: substrates, differential diagnosis and management. 3rd ed. St. Louis: Mosby; 2013.
Google Scholar
Morgan AT, Masterton R, Pigdon L, Connelly A, Liegeois FJ. Functional magnetic resonance imaging of chronic dysarthric speech after childhood brain injury: reliance on a left-hemisphere compensatory network. Brain. 2013;136:646–57.
McLeod S, Crowe K, Shahaeian A. Intelligibility in context scale: normative and validation data for English-speaking preschoolers. Lang, speech, hearing Serv Sch. 2015;46:266–76.
Bishop DV. Children’s communication checklist, Second Edition. 2nd ed. London: Pearson; 2003.
Fenson L, Marchman VA, Thal DJ, Dale PS, Reznick S, Bates E. The MacArthur-Bates communicative development inventories user’s guide and technical manual. 2nd ed. Baltimore: Brookes; 2006.
Wetherby AM, Prizant BM. Communication and Symbolic Behavior Scales Developmental Profile™ Infant-Toddler Checklist. Baltimore: Brookes; 2002.
Sparrow SS, Cicchetti DV, Saulnier CA. Vineland Adaptive Behaviour Scales, third edition. 3rd ed. Bloomington: Pearson; 2016.
Morgan AT, Fisher SE, Scheffer IE, Hildebrand MS. FOXP2-Related Speech and Language Disorders. In: GeneReviews(R) [Internet]. Seattle: University of Washington; 2017. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved
Chenausky KV, Brignell A, Morgan A, Gagne D, Norton A, Tager-Flusberg H, et al. Factor analysis of signs of childhood apraxia of speech. J Commun Disord. 2020:106033. https://doi.org/10.1016/j.jcomdis.2020.106033 .
Morgan AT, Murray E, Liegeois FJ. Interventions for childhood apraxia of speech. Cochrane Database Syst Rev. 2018;5:CD006278. https://doi.org/10.1002/14651858.CD006278.pub3 .
Dereu M, Roeyers H, Raymaekers R, Meirsschaut M, Warreyn P. How useful are screening instruments for toddlers to predict outcome at age 4? General development, language skills, and symptom severity in children with a false positive screen for autism spectrum disorder. Eur Child Adolesc Psychiatry. 2012;21:541–51.
Fujiki M, Brinton B, Hart CH, Olsen J, Coombs M. Using measurement invariance to study social withdrawal in children with developmental language disorders. Lang, Speech, Hear Serv Sch. 2019;50:253–66.
Durkin K, Toseeb U, Botting N, Pickles A, Conti-Ramsden G. Social confidence in early adulthood among young people with and without a history of language impairment. J Speech Lang Hear Res. 2017;60:1635–47.
Coccaro N, Tota G, Zagaria A, Anelli L, Specchia G, Albano F. SETBP1 dysregulation in congenital disorders and myeloid neoplasms. Oncotarget. 2017;8:51920–35.
Minakuchi M, Kakazu N, Gorrin-Rivas MJ, Abe T, Copeland TD, Ueda K, et al. Identification and characterization of SEB, a novel protein that binds to the acute undifferentiated leukemia-associated protein SET. Eur J Biochem. 2001;268:1340–51.
Piazza R, Magistroni V, Redaelli S, Mauri M, Massimino L, Sessa A, et al. SETBP1 induces transcription of a network of development genes by acting as an epigenetic hub. Nat Commun. 2018;9:2192.
Piazza R, Valletta S, Winkelmann N, Redaelli S, Spinelli R, Pirola A, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet. 2013;45:18–24.
Download references
Acknowledgements
Sincere thanks to the children, families and clinicians who took part in this project. A special thanks to Haley Oyler, president of the SETBP1 Society for assistance with recruitment and for providing unique parental insights into children with SETBP1 haploinsufficiency disorder.
Funding was provided by National Health and Medical Research Council (NHMRC) Practitioner Fellowship #1105008 (AM); NHMRC Investigator Grant #1195955. NHMRC Centre of Research Excellence in Speech and Language Neurobiology #1116976 (AM, DA, SEF); and the Max Planck Society (MMKW, SEF). This work was supported by the Victorian Government’s Operational Infrastructure Support Program.
Author information
These authors contributed equally: Angela Morgan, Ruth Braden
Authors and Affiliations
Speech & Language, Murdoch Children’s Research Institute, Melbourne, Victoria, Australia
Angela Morgan, Ruth Braden & David Amor
Department of Paediatrics, University of Melbourne, Parkville, Victoria, Australia
Angela Morgan & Ruth Braden
Royal Children’s Hospital, Melbourne, Victoria, Australia
Angela Morgan & David Amor
Victorian Clinical Genetics Service, Melbourne, Victoria, Australia
Department of Audiology and Speech Pathology, University of Melbourne, Parkville, Victoria, Australia
Language and Genetics Department, Max Planck Institute for Psycholinguistics, Nijmegen, The Netherlands
Maggie M. K. Wong & Simon E. Fisher
Service de Génétique Médicale, Centre Hospitalier Universitaire d’Angers, Angers, France
Estelle Colin
UCL Great Ormond Street Institute of Child Health, London, UK
Frederique Liégeois
Boston Children’s, Harvard Medical Centre, Boston, MA, USA
Siddharth Srivastava
Centre for Neuroscience of Speech, Department of Audiology and Speech Pathology, University of Melbourne, Melbourne, Victoria, Australia
Service de Génétique, Centre Hospitalier Universitaire Caen Normandie, Caen, France
Varoona Bizaoui & Kara Ranguin
Donders Institute for Brain, Cognition and Behaviour, Radboud University, Nijmegen, The Netherlands
Simon E. Fisher
Radboud University Medical centre, Nijmegen, The Netherlands
Bregje W. van Bon
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Angela Morgan .
Ethics declarations
Conflict of interest.
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Reprints and permissions
About this article
Cite this article.
Morgan, A., Braden, R., Wong, M.M.K. et al. Speech and language deficits are central to SETBP1 haploinsufficiency disorder. Eur J Hum Genet 29 , 1216–1225 (2021). https://doi.org/10.1038/s41431-021-00894-x
Download citation
Received : 20 November 2020
Revised : 26 March 2021
Accepted : 06 April 2021
Published : 27 April 2021
Issue Date : August 2021
DOI : https://doi.org/10.1038/s41431-021-00894-x
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies


Am Fam Physician. 2024;109(4):361-362
As published by the USPSTF.
The full recommendation statement is available at https://www.uspreventiveservicestaskforce.org/uspstf/recommendation/speech-and-language-delay-and-disorders-in-children-age-5-and-younger-screening .
The USPSTF recommendations are independent of the U.S. government. They do not represent the views of the Agency for Healthcare Research and Quality, the U.S. Department of Health and Human Services, or the U.S. Public Health Service.
This series is coordinated by Joanna Drowos, DO, contributing editor.
A collection of USPSTF recommendation statements published in AFP is available at https://www.aafp.org/afp/uspstf .
Continue Reading

More in AFP
Copyright © 2024 by the American Academy of Family Physicians.
This content is owned by the AAFP. A person viewing it online may make one printout of the material and may use that printout only for his or her personal, non-commercial reference. This material may not otherwise be downloaded, copied, printed, stored, transmitted or reproduced in any medium, whether now known or later invented, except as authorized in writing by the AAFP. See permissions for copyright questions and/or permission requests.
Copyright © 2024 American Academy of Family Physicians. All Rights Reserved.
Princeton University
Princeton engineering, can language models read the genome this one decoded mrna to make better vaccines..
By Scott Lyon
April 8, 2024

Princeton researchers led by Mengdi Wang have developed a language model to home in on partial genome sequences and optimize those sequences to improve function for the development of mRNA vaccines and other therapies. Illustration from Adobe Stock.
The same class of artificial intelligence that made headlines coding software and passing the bar exam has learned to read a different kind of text — the genetic code.
That code contains instructions for all of life’s functions and follows rules not unlike those that govern human languages. Each sequence in a genome adheres to an intricate grammar and syntax, the structures that give rise to meaning. Just as changing a few words can radically alter the impact of a sentence, small variations in a biological sequence can make a huge difference in the forms that sequence encodes.
Now Princeton University researchers led by machine learning expert Mengdi Wang are using language models to home in on partial genome sequences and optimize those sequences to study biology and improve medicine. And they are already underway.
In a paper published April 5 in the journal Nature Machine Intelligence, the authors detail a language model that used its powers of semantic representation to design a more effective mRNA vaccine such as those used to protect against COVID-19.
Found in Translation

Scientists have a simple way to summarize the flow of genetic information. They call it the central dogma of biology. Information moves from DNA to RNA to proteins. Proteins create the structures and functions of living cells.
Messenger RNA, or mRNA, converts the information into proteins in that final step, called translation. But mRNA is interesting. Only part of it holds the code for the protein. The rest is not translated but controls vital aspects of the translation process.
Governing the efficiency of protein production is a key mechanism by which mRNA vaccines work. The researchers focused their language model there, on the untranslated region, to see how they could optimize efficiency and improve vaccines.
After training the model on a small variety of species, the researchers generated hundreds of new optimized sequences and validated those results through lab experiments. The best sequences outperformed several leading benchmarks for vaccine development, including a 33% increase in the overall efficiency of protein production.
Increasing protein production efficiency by even a small amount provides a major boost for emerging therapeutics, according to the researchers. Beyond COVID-19, mRNA vaccines promise to protect against many infectious diseases and cancers.
Wang, a professor of electrical and computer engineering and the principal investigator in this study, said the model’s success also pointed to a more fundamental possibility. Trained on mRNA from a handful of species, it was able to decode nucleotide sequences and reveal something new about gene regulation. Scientists believe gene regulation, one of life’s most basic functions, holds the key to unlocking the origins of disease and disorder. Language models like this one could provide a new way to probe.
Wang’s collaborators include researchers from the biotech firm RVAC Medicines as well as the Stanford University School of Medicine.
The Language of Disease
The new model differs in degree, not kind, from the large language models that power today’s AI chat bots. Instead of being trained on billions of pages of text from the internet, their model was trained on a few hundred thousand sequences. The model also was trained to incorporate additional knowledge about the production of proteins, including structural and energy-related information.
The research team used the trained model to create a library of 211 new sequences. Each was optimized for a desired function, primarily an increase in the efficiency of translation. Those proteins, like the spike protein targeted by COVID-19 vaccines, drive the immune response to infectious disease.
Previous studies have created language models to decode various biological sequences, including proteins and DNA, but this was the first language model to focus on the untranslated region of mRNA. In addition to a boost in overall efficiency, it was also able to predict how well a sequence would perform at a variety of related tasks.
Wang said the real challenge in creating this language model was in understanding the full context of the available data. Training a model requires not only the raw data with all its features but also the downstream consequences of those features. If a program is designed to filter spam from email, each email it trains on would be labeled “spam” or “not spam.” Along the way, the model develops semantic representations that allow it to determine what sequences of words indicate a “spam” label. Therein lies the meaning.
Wang said looking at one narrow dataset and developing a model around it was not enough to be useful for life scientists. She needed to do something new. Because this model was working at the leading edge of biological understanding, the data she found was all over the place.
“Part of my dataset comes from a study where there are measures for efficiency,” Wang said. “Another part of my dataset comes from another study [that] measured expression levels. We also collected unannotated data from multiple resources.” Organizing those parts into one coherent and robust whole — a multifaceted dataset that she could use to train a sophisticated language model — was a massive challenge.
“Training a model is not only about putting together all those sequences, but also putting together sequences with the labels that have been collected so far. This had never been done before.”
The paper, “A 5′ UTR Language Model for Decoding Untranslated Regions of mRNA and Function Predictions,” was published in Nature Machine Learning. Additional authors include Dan Yu, Yupeng Li, Yue Shen and Jason Zhang, from RVAC Medicines; Le Cong from Stanford; and Yanyi Chu and Kaixuan Huang from Princeton.
Related News

Episode 2: That Magic Touch

Episode 3: Haydn Seek

Episode 1: Stanley Jordan Pulls Out All the Stops

Personalizing ChatGPT can make it more offensive, researchers find

Princeton IP Accelerator funding awarded to support promising new technologies

Built for AI, this chip moves beyond transistors for huge computational gains

Mengdi Wang
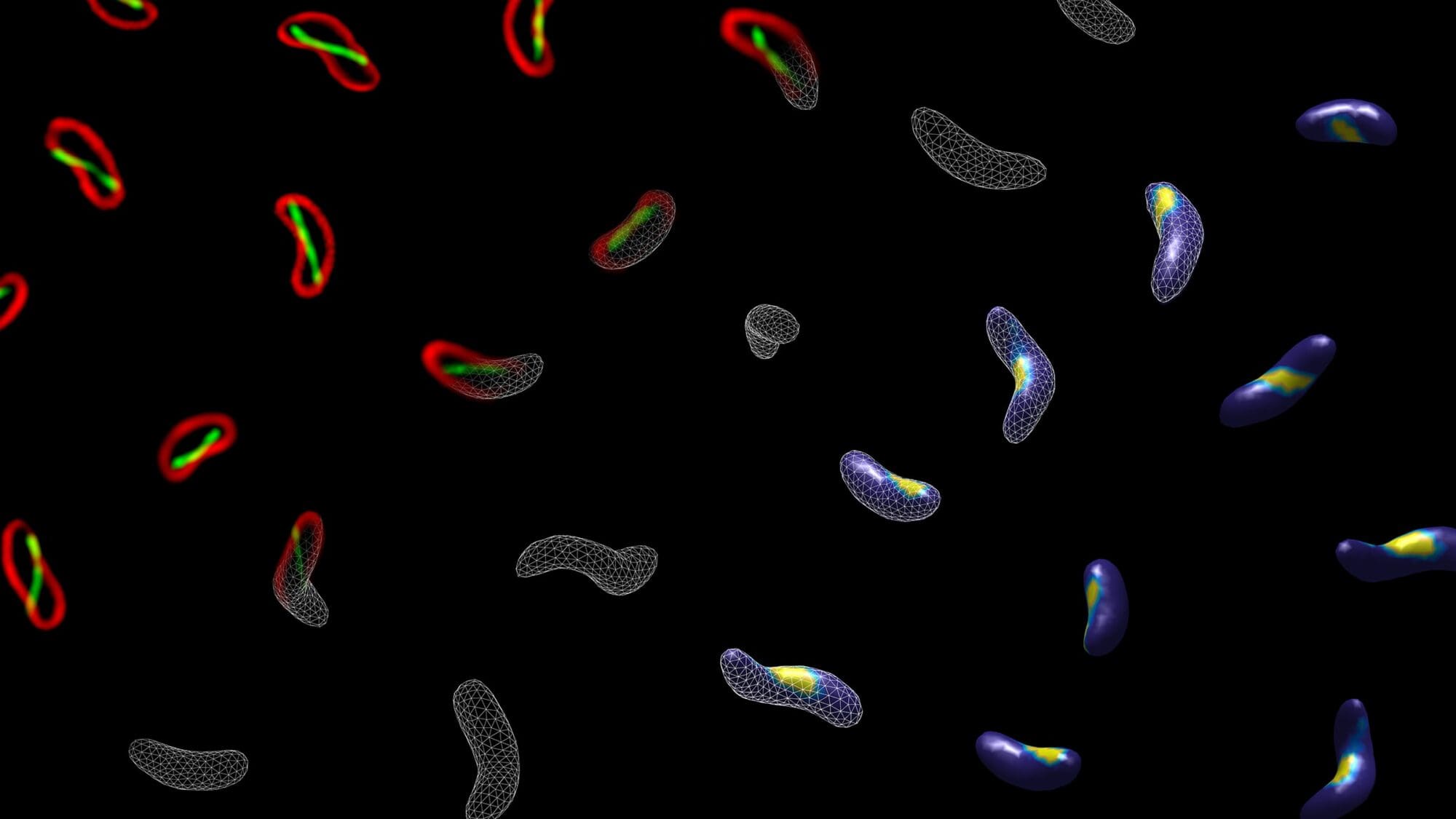
Bioengineering and Health

Data Science
Related departments and centers.

Electrical and Computer Engineering

Omenn-Darling Bioengineering Institute
Study of the binding of nuclear proteins from Plasmodium berghei strains with different chloroquine sensitivity to oligonucleotides corresponding to regulatory elements of the multidrug resistance gene
- Experimental Studies
- Published: 03 April 2008
- Volume 2 , pages 94–100, ( 2008 )
Cite this article
- T. G. Pankova 1 ,
- T. M. Igonina 1 ,
- V. F. Kobzev 1 &
- T. I. Merkulova 1
20 Accesses
Explore all metrics
Using electrophoretic mobility shift assay the proteins, specifically bound to the double-stranded oligonucleotides corresponding the binding sites of the transcription factors of AP1 family, NF-IL6 and SP1 involved in the up-regulation of human multidrug resistance (mdr1) gene and also proteins bound to the oligonucleotide corresponding to the element responsive for the stimulation by serum (SRE) have been found in the nuclear extracts of the rodent malaria parasite Plasmodium berghei ( P. berghei ). P. berghei strains exhibiting different chloroquine sensitivity are characterized by different patterns of nuclear protein binding to the oligonucleotides used. Mutations in the consensus sequences of AP1, NF-IL6, and SRE caused impairments in binding of some proteins; this suggests the existence of malaria parasite nuclear proteins containing DNA binding domains, which share similarity with corresponding DNA binding domains of transcription factors NF-IL6, SRF1 and the members of AP1 family. The results obtained suggest profound alterations in the regulatory apparatus of plasmodium during its selection for chloroquine resistance.
This is a preview of subscription content, log in via an institution to check access.
Access this article
Price includes VAT (Russian Federation)
Instant access to the full article PDF.
Rent this article via DeepDyve
Institutional subscriptions
Similar content being viewed by others
Biochemical and functional characterization of plasmodium falciparum dna polymerase δ.
Jitlada Vasuvat, Atcha Montree, … Porntip Chavalitshewinkoon-Petmitr

Inhibitory effects of anthracyclines on partially purified 5′–3′ DNA helicase of Plasmodium falciparum
Pongruj Rattaprasert, Pattra Suntornthiticharoen, … Porntip Chavalitshewinkoon-Petmitr

Molecular characterization of Plasmodium falciparum uracil-DNA glycosylase and its potential as a new anti-malarial drug target
Thidarat Suksangpleng, Ubolsree Leartsakulpanich, … Porntip Chavalitshewinkoon-Petmitr
Wellems, T.E., Science , 2002, vol. 298, pp. 124–126.
Article CAS Google Scholar
Salganik, R.I., Pankova, T.G., Chekhonadskikh, T.V., and Igonina, T.M., Bull. WHO , 1987, vol. 65, pp. 381386.
Google Scholar
Ndifor, A.M., Howells, R.E., Bray, P.G., Ngu, J.L., and Ward, S.A., Antimicrob. Agents and Chemother , 1993, vol. 37, pp. 1318–1323.
Pankova, T.G., Igonina, T.M., Mayer, T.V., and Salganik, R.I., Mol. Biol. (Moscow) , 1996, vol. 30, 43–50.
CAS Google Scholar
Padua, R.A., Exp. Parasitology , 1981, vol. 52, pp. 419426.
Article Google Scholar
Foote, S.J., Kyle, D.E., Martin, R.K., Oduola, A.M.J., Kemp, D.J., and Cowman, A.F., Nature , 1990, vol. 345, pp. 255–258.
Sidhu, A.B., Verdier-Pinard, D., and Fidock, D.A., Science , 2002, vol. 298, pp. 210–213.
Reed, M.B., Saliba, K.J., Caruana, S.R., Kirk, K., and Cowman, A.F., Nature , 2000, vol. 403, pp. 906–909.
Bray, P.G., Martin, R.E., Tilley, L., Ward, S.A., Kirk, K., and Fidock, D.A., Mol. Microbiol. , 2005, vol. 56, pp. 323–333.
Ekong, R.M., Robson, K.J.H., Baker, D.A., and Warhurst, D.C., Parasitology , 1992, vol. 106, pp. 107–125.
Myrick, A., Munasinghe, A., Patankar, S., and Wirth, D.F., Mol. Microbiol. , 2003, vol. 49, pp. 671–683.
Waller, K.L., Muhle, R.A., Ursos, L.M., Horrocks, P., Verdier-Pinard, D., Sidhu, A.B., Fujioka, H., Roepe, PD., and Fidock, D.A., J. Biol. Chem. , 2003, vol. 278, pp. 33 593–33 601.
Gunasekera, A.M., Patankar, S., Schug, J., Eisen, G., and Wirth, D.F., Mol. Microbiol. , 2003, vol. 50, pp. 1229–1239.
Daschner, Ph.J., Ciolino, H.P., Plouzek, C.A., and Yeh, G.C., Breast Cancer Res. Treat. , 1999, vol. 53, pp. 229–240.
Combates, N.J., Rzepka, R.W., Chen, Y N.P., and Cohen, D., J. Biol. Chem. , 1994, vol. 269, pp. 29715–29719.
Cornwell, M.M. and Smith, D.E., J. Biol. Chem. , 1993, vol. 268, pp. 19505–19511.
Rabinovich, S.A., Kulikovskaya, I.M., Maksakovskaya, E.V., Chekhonadskikh, T.V., Pankova, T.G., and Salganik, R.I., Bull. WHO , 1987, vol. 65, pp. 387–389.
Francois, G., Bringmann, G., Dochez, C., Schneider, C., Timperman, G., and AkeAssi, L., J. Ethnopharmacol. , 1995, vol. 46, pp. 115–120.
Horrocks, P and Lanzer, M., Mol. Biochem. Parasitol. , 1999, vol. 99, pp. 77–87.
Vasil’ev, G.V., Merkulov, V.M., Kobzev, VF., Merkulova, T.I., Ponomarenko, M.P., Podkolodnaya, O.F., Ponomarenko, Yu.V., and Kolchanov, N.A., Mol. Biol. (Moscow) , 2000, vol. 34, pp. 214–222.
Treisman, R., Trends Biochem. Sci. , 1992, vol. 17, pp. 423–426.
Llinas, M., Bozdech, Z., Wong, E.D., Adai, A.T., and DeRisi, J.L., Nucleic Acids Res. , 2006, vol. 34, pp. 1166–1173.
Wilson, R.J.M., BioEssays , 2004, vol. 26}, pp. 339–342.
Coulson, R.M.R., Hall, N., and Ouzounis, C.A., Genome Res. , 2004, vol. 14, pp. 1548–1554.
Noort, V. and Huynen, M.A., Orends in Genetics , 2006, vol. 22, pp. 73–78.
Download references
Author information
Authors and affiliations.
Institute of Cytology and Genetics, Siberian Branch, Russian Academy of Sciences, pr. Akademika Lavrent’eva 10, Novosibirsk, 630090, Russia
T. G. Pankova, T. M. Igonina, V. F. Kobzev & T. I. Merkulova
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to T. G. Pankova .
Additional information
Original Russian Text © T.G. Pankova, T.M. Igonina, V.F. Kobzev, T.I. Merkulova, 2008, published in Biomeditsinskaya Khimiya.
Rights and permissions
Reprints and permissions
About this article
Pankova, T.G., Igonina, T.M., Kobzev, V.F. et al. Study of the binding of nuclear proteins from Plasmodium berghei strains with different chloroquine sensitivity to oligonucleotides corresponding to regulatory elements of the multidrug resistance gene. Biochem. Moscow Suppl. Ser. B 2 , 94–100 (2008). https://doi.org/10.1007/s11828-008-1011-2
Download citation
Received : 02 March 2007
Published : 03 April 2008
Issue Date : March 2008
DOI : https://doi.org/10.1007/s11828-008-1011-2
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- chloroquine resistance
- transcription factors
- oligodesoxyribonucleotides
- electrophoretic mobility shift assay
- Find a journal
- Publish with us
- Track your research

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
Evolution of language: Lessons from the genome
Simon e. fisher.
1 Language and Genetics Department, Max Planck Institute for Psycholinguistics, Wundtlaan 1, 6525 XD Nijmegen, The Netherlands
2 Donders Institute for Brain, Cognition and Behaviour, Radboud University, 6525 EN Nijmegen, The Netherlands
The post-genomic era is an exciting time for researchers interested in the biology of speech and language. Substantive advances in molecular methodologies have opened up entire vistas of investigation that were not previously possible, or in some cases even imagined. Speculations concerning the origins of human cognitive traits are being transformed into empirically addressable questions, generating specific hypotheses that can be explicitly tested using data collected from both the natural world and experimental settings. In this article, I discuss a number of promising lines of research in this area. For example, the field has begun to identify genes implicated in speech and language skills, including not just disorders but also the normal range of abilities. Such genes provide powerful entry points for gaining insights into neural bases and evolutionary origins, using sophisticated experimental tools from molecular neuroscience and developmental neurobiology. At the same time, sequencing of ancient hominin genomes is giving us an unprecedented view of the molecular genetic changes that have occurred during the evolution of our species. Synthesis of data from these complementary sources offers an opportunity to robustly evaluate alternative accounts of language evolution. Of course, this endeavour remains challenging on many fronts, as I also highlight in the article. Nonetheless, such an integrated approach holds great potential for untangling the complexities of the capacities that make us human.
Our speech and language capacities enable us to acquire vocabularies of many thousands of words, assemble them into a myriad of structured meaningful expressions, and convey thoughts to others by mapping meaning to sound, and back again. In the twenty-first century, we are witnessing dramatic advances in deciphering the genetic architecture underlying these fascinating aspects of the human condition. By directly borrowing state-of-the-art gene mapping approaches from studies of typical biomedical traits, and applying them to scientific studies of language for the first time, it has become feasible to start tracing out relevant genetic networks (Graham & Fisher, 2015 ). The language sciences are thereby witnessing a paradigm shift, moving far beyond prior speculative models in which genes have been invoked as abstract entities with mysterious powers, an issue that I have discussed in detail elsewhere (Fisher, 2006 ; Fisher & Vernes, 2015 ). Gene discovery strategies take advantage of the modern human population as a kind of natural experiment (e.g. Narasimhan et al., 2016 ) for uncovering potential connections between genotype (the genetic constitution of an individual at a particular locus or set of loci) and phenotype (the appearance of that individual in terms of a particular characteristic, be it physical, biochemical, physiological, behavioural, etc.). Applying this framework to the language sciences entails searching for statistically significant correlations between variations observed at the genomic level and variability in speech- and language-related skills, with the aim of establishing causal relationships.
Due to rapid progress in molecular methods, we now have a particularly comprehensive view of the ways that genes and genomes vary from one person to the next (see The 1000 Genomes Project Consortium, 2015 ). Genetic variations range from mutations that are extremely rare (perhaps even unique to one individual or family) all the way through to common variants that are found in populations at high frequency. Rare mutations can have severe effects on gene products, for instance by preventing an important protein from being made or interfering with its function, and could thereby be sufficient to cause a major disorder affecting one or more tissues of the body. Common variants (also known as polymorphisms) tend to have much more subtle effects on gene function, for example by leading to a slight change in the quantity or activity of the protein that a gene codes for. Indeed, many genetic polymorphisms are completely benign, and those that do have biological effects typically show probabilistic relationships with phenotypic outcomes, for example by partially altering the risk of a particular disease or accounting for a tiny difference in a quantitatively defined trait (height, blood pressure, body-mass index, and so on). Thus, a person’s phenotype can be considered the consequence of combinatorial effects of all the rare and common gene variants that their genome carries, together with their interactions with the environment, as well as stochastic factors. In contrast to the current wealth of information on genotypic variability in human populations, we know considerably less about the nature of phenotypic variability in speech and language skills. (Perhaps this is in part due to the emphasis that linguists have traditionally placed on universals.) In principle, searching for genotype-phenotype correlations is an approach that can be applied across the entire spectrum of variability observed in modern humans, but, thus far, most emphasis has been placed on studies of pathology (Newbury & Monaco, 2010 ).
Opening molecular windows
A major focus of the field has concerned neurodevelopmental disorders in which children suffer from disproportionate impairments in mastering aspects of speech and language, against a background of relatively preserved cognitive function, and despite adequate exposure to language input in their environment (Bishop, 2001 ). It has long been known that such unexplained disorders of speech and language development tend to cluster in families, and are highly heritable (that is, a substantial proportion of risk is due to genetic factors). Studies of DNA samples from families with rare forms of these disorders have allowed geneticists to go further and pinpoint the specific genetic disruptions that are causal (Graham, Deriziotis, & Fisher, 2015 ). The most often cited example is the identification of a FOXP2 mutation causing speech apraxia, along with expressive and receptive language impairments, in multiple generations of a large British Caucasian family, referred to in the literature as the KE family (Lai, Fisher, Hurst, Vargha-Khadem, & Monaco 2001 ). However, this is certainly not the only mutation to have been clearly implicated in this type of disorder. Additional families and unrelated cases have been found carrying different causative mutations of FOXP2 , and rare disruptions in other genes, such as ERC1 and BCL11A , have been reported in children with a similar profile of severe speech and language problems (see Graham & Fisher, 2015 for a detailed overview of the latest findings). In addition, studies of common genetic variation have begun to identify polymorphisms that may make more subtle contributions to language pathology; for example, putative risk variants of the CNTNAP2 gene have been associated with reduced performance on language tasks in children with specific language impairment (Vernes et al., 2008 ).
In studying genetic underpinnings of speech and language capacities, the successful isolation of a gene that contributes to the relevant phenotype is not the endgame. To the contrary, it is a starting point—the value of implicating a specific gene in a trait lies in the novel avenues of investigation that can be followed as a consequence. The identification of a gene like FOXP2 opens up unique molecular windows into both the neural bases and the evolutionary origins of speech and language (Fisher & Scharff, 2009 ; Scharff & Petri, 2011 ). I will consider each of these lines of research in turn below.
Insights into neural pathways
Regarding neural bases, an array of sophisticated experimental approaches, ranging from analyses of human neurons grown in a dish (Vernes et al., 2007 ), through to studies of circuits and behaviour in genetically manipulated animal models (French & Fisher, 2014 ; Wohlgemuth, Adam, & Scharff, 2014 ), can reveal fundamental roles of the gene of interest in brain development and function. These methods may appear relatively new for scientists studying language, but they are established mainstays of molecular neuroscience, developmental neurobiology and other related fields. Indeed, some language scientists may be surprised to learn that the general principles governing how genetic programs help build a complex nervous system are already well worked out (reviewed by Fisher & Vernes, 2015 ). The products of genes (RNA molecules and proteins) interact with each other to mediate the proliferation of cells that will become neurons, their differentiation into particular types of neurons, and migration of these neurons during development to their final locations in the brain (Tan & Shi, 2013 ). Moreover, connectivity patterns in the central nervous system emerge from a tight interplay of genetic and environmental factors—gene products underlie mechanisms by which projections emerge from the neuronal cell body to become dendrites and axons, as well as the growth and guidance of axons towards their target neurons (Kolodkin & Tessier-Lavigne, 2011 ). Even the strengthening or weakening of the individual connections between neurons (synapses), the basis of learning and memory, depends critically on activities of certain sets of genes (Holtmaat & Svoboda, 2009 ).
Having pinpointed a gene involved in a language-related disorder, purely based on genomic data from families and cases, researchers can then delve deeply into the functional correlates to uncover how the implicated gene impacts on neuronal proliferation, differentiation, connectivity, plasticity, and so on, drawing from the growing set of elegant experimental tools and systems that molecular neuroscience has to offer (Fisher & Vernes, 2015 ). In addition, the mutations that yield speech and language impairments can be directly introduced into cells grown in the laboratory, or into animal models, to help understand how the crucial mechanisms and pathways go awry in disorder. For example, the FOXP2 mutation that causes a severe speech and language disorder in the KE family is a change to a single letter of DNA, leading to alteration of the amino-acid sequence of the encoded protein. Genetic engineering makes it possible to create and study human neurons that carry this same change, or even insert the identical mutation into another species, such as a mouse, an issue I return to later.
Before moving on, three important take-home messages from molecular neurobiology are worth emphasising. First, clearly a gene does not itself specify a particular behaviour output, nor does it even specify a particular neural circuit. The pathways by which molecular factors impact on neural circuitry and cognitive functions are by their very nature indirect and must occur via intermediate effects on the types of neurobiological processes discussed above (proliferation, differentiation, connectivity, plasticity, etc.). The necessarily complex mappings from gene to behaviour mean that discussions that centre on an abstract “gene for language” are unconstructive; more nuanced accounts built on biological principles give an opportunity for real progress (Fisher, 2006 ). Second, typically a gene does not have a single restricted function, but instead contributes to more than one process, is active in a range of distinct cell-types, and/or plays roles at multiple developmental time points or in different environmental contexts. This widespread property of gene action is usually referred to by the technical term of pleiotropy . The same gene can thus have multiple roles within the brain, as well as contributing to development and function of non-neural tissues. Given that the human genome comprises only ~20,000 protein-coding genes, it is perhaps unsurprising that each gene is “re-used” in a number of different contexts in the brain and body, with the precise functions of the encoded protein depending on the other proteins that are active in the tissue. This leads to the third take-home message, which is that genes and proteins do not act in isolation but interact with each other in networks and complexes. Indeed, the combinatorial nature of gene activity is a highly valuable feature for researchers interested in deciphering the biology underlying a trait of interest. When FOXP2 was first identified, it quickly became clear that this gene encoded a type of protein that, working together with other related proteins, regulates how certain sets of genes (downstream targets) are switched on and off. In other words, FOXP2 represents a hub in a genetic network. The tools of molecular biology have since enabled the identification of additional elements of this network, including interactors, like FOXP1 and TBR1 , and downstream targets, such as CNTNAP2 , that are also implicated in neurodevelopmental disorders and language-related traits (Deriziotis et al., 2014 ; Sollis et al., 2016 ; Vernes et al., 2008 ). In general, as our understanding of gene networks becomes more and more sophisticated, this holds great promise for helping to successfully bridge the gap between the genome and the brain.
Towards empirical studies of evolution
Identification of genes implicated in speech and language disorder also provides valuable entry points for empirical studies of evolutionary origins of human traits, rooted in biological data (Enard, 2011 ; Fisher & Marcus, 2006 ). By taking the DNA sequence of a known language-related gene (and the amino-acid sequence of its protein product) and comparing it to corresponding sequences found in different species across the animal kingdom, it is possible to reconstruct the likely evolutionary history of the gene, determining a time window when it first emerged, the nature of alterations along different lineages, and whether it has shown a distinctive profile of change in our most recent ancestors. (To be clear, here I focus on what we can learn from comparative analyses of extant species, and address the promise of ancient hominin data in a later section of this article.) It is also feasible, although more difficult, to characterize where and when the gene product is active in different structures of the developing and adult human brain (based on analyses of post-mortem tissue), and compare this neural expression pattern to that seen in other species. If potentially significant evolutionary differences are detected in a gene of interest, this can yield hypotheses about functional impact that can be empirically evaluated using model systems (Enard 2014 ).
Again, studies of FOXP2 give a nice illustration of the concept. Comparative analyses revealed that, far from being unique to humans, this gene has a deep evolutionary history and is present in similar form in distantly related vertebrate species (Scharff & Petri, 2011 ). Conservation has been found not only at the DNA/protein sequence level but also in assessments of neural expression patterns (e.g. Bonkowsky & Chien, 2005 ; Haesler et al., 2004 ; Kato et al., 2014 ; Lai, Gerrelli, Monaco, Fisher, & Copp, 2003 ; Teramitsu, Kudo, London, Geschwind, & White, 2004 ), with findings also supported by experiments assessing gene function in several different species (French & Fisher, 2014 ; Wohlgemuth, Adam, & Scharff, 2014 ). Such data suggest that the gene has ancient roles in the development and function of certain brain circuits involving the cortex, basal ganglia and cerebellum, with relevance for sensorimotor integration and motor-skill learning (e.g. French et al., 2012 ; Groszer et al., 2008 ; Haesler et al., 2007 ; Murugan, Harward, Scharff, & Mooney, 2013 ). Against this background of high evolutionary conservation, the human version of FOXP2 carries two amino-acid coding changes that occurred after splitting from the common ancestor with the chimpanzee, leading to a specific hypothesis that one or both of the evolutionary changes might have been important for the emergence of speech and language on our lineage (Enard et al., 2002 ). Crucially, researchers have gone on to test this hypothesis using the same model systems and experimental approaches that are used for investigating mutations that cause disorder. For example, when the key evolutionary amino-acid changes were inserted into genetically modified mice, the mice showed higher levels of plasticity of synapses in cortico-basal ganglia circuits (reviewed by Enard, 2011 ). By contrast, when mice were genetically modified to carry a disruptive FOXP2 mutation that is known to cause a speech and language disorder (the mutation from the KE family) such mice showed lower levels of synaptic plasticity in cortico-basal ganglia circuits, consistent with a loss of function (see Groszer et al., 2008 ). Thus, it seems that the experiments with mice carrying the evolutionary changes are capturing something about the biological significance of those changes, rather than simply reflecting disturbance of existing pathways. Investigations of evolutionary history have also been used to assess recent positive selection of broader networks regulated by FOXP2 (Ayub et al., 2013 ) and to evaluate other candidate genes implicated in language-related phenotypes, such as KIAA0319 , ROBO1 , ROBO2 , and CNTNAP2 (Mozzi et al., 2016 ).
Learning from our genomes
The accumulated data from molecular studies support the view that genetic underpinnings of speech and language skills are highly multifactorial, indicating that no single locus is sufficient by itself to account for such traits (Graham & Fisher, 2015 ). In particular, the genes that have been most clearly implicated in relevant developmental disorders can explain only a small subset of affected families and cases. The majority of discoveries have thus far depended on laborious detective work using a standard genetics toolkit, along with some serendipity in targeting unusual families and cases with monogenic forms of disorder. However, the advent of next generation DNA sequencing means that we can now sequence the whole genome (or at least a substantial proportion of it) in any human individual for less than 1000 euros, in a matter of days, and requiring only a sample of saliva. This development holds considerable promise for increasing our knowledge of the genetic aetiology of rare forms of speech and language disorder. Already, sequencing of the entire coding parts of the genome (the exome) is beginning to make its mark on the field (e.g. Villanueva et al., 2015 ). At the same time, even cheaper methods enable rapid high-throughput screening of hundreds of thousands of common genetic variations for less than 100 euros per person, allowing for large-scale genome-wide studies of common forms of language-related disorders (Gialluisi et al., 2014 ) and even investigations of normal variation in the general population (Luciano et al., 2013 ; St Pourcain et al., 2014 ). It will also be interesting to study people at the other extreme of the phenotypic spectrum, such as those rare individuals who have exceptional abilities to master many languages. Of course, the availability of inexpensive accessible techniques for capturing genomic variation leads to its own new challenges, primarily in distilling biologically meaningful signals from vast datasets, but creative solutions are in place, or on the horizon (see Graham & Fisher, 2015 for further discussion). These efforts will yield further candidate genes and associated networks for targeted analyses of neural function and evolutionary history.
Insights from ancient hominin DNA
Advances in genomics are not only transforming gene-mapping studies of modern day humans. In one of the most astonishing technological achievements of molecular biology, it is now possible to read off sequences of nuclear DNA from ancient organisms that are extinct, giving an unprecedented glimpse into the genomes of the past (Pääbo, 2014 ). Molecular anthropologists have successfully sequenced entire genomes of ancient hominins using archaeological samples estimated to be tens of thousands of years old (Fu et al., 2014 ; Meyer et al., 2012 ; Prüfer et al., 2014 ). Sequenced genomes are available not only for ancestors on our own lineage, such as a ~45,000-year-old modern human (Fu et al., 2014 ), but also for >50,000-year-old bones from independent hominin branches, including Neandertals (Prüfer et al., 2014 ) and Denisovans (Meyer et al., 2012 ). Note that, although the samples themselves are tens of thousands of years old, the time-depth they provide for comparative evolutionary analyses is much greater, because the common ancestor of modern humans and Neandertal/Denisovan hominins existed several hundred thousand years ago. Most recently, nuclear DNA sequences have been recovered from two hominin samples that are estimated to be >430,000 years old, although DNA of this age is much too degraded to ever yield a complete genome sequence (Meyer et al., 2016 ).
Ancient hominin DNA sequences, together with matching data from extant primates, provide invaluable additional datapoints for evaluating the evolutionary significance of changes in language-related genes. As before, FOXP2 provides an interesting case in point. Examination of the two amino-acid coding changes that distinguish the human sequence from that of chimpanzees reveals that they are not unique to humans (Krause et al. 2007 ) but also present in Neandertal and Denisovan samples. Some have taken this as one of the supporting points in favour of the view that our Neandertal cousins also possessed some form of spoken language (Dediu & Levinson, 2013 ), although it is important to stress that the status of a single gene is not enough to resolve whether or not an ancient hominin could speak. Further in-depth comparisons of modern human and Neandertal versions of FOXP2 , examining the parts of the genetic locus that do not code for protein, identified human-specific changes that might potentially affect the way that the gene is regulated (Maricic et al., 2013 ). Although the functional data to support this hypothesis are tentative at present (Maricic et al., 2013 ), this work represents another example where ideas about evolutionary impact do not remain speculation, but are open to empirical testing.
Weighing up alternatives
Importantly, the availability of virtually complete genome sequences for modern and ancestral humans, Neandertals, Denisovans and great apes, allows molecular anthropologists to generate genome-wide catalogues of evolutionary changes that occurred on the different lineages during distinct evolutionary periods (Pääbo, 2014 ). These comparative molecular catalogues are of enormous value for both constraining and enhancing accounts of the origins of human traits, including (but not limited to) our linguistic capacities. For example, certain hypotheses concerning the origins of language posit the existence of just a single causative DNA mutation, occurring on the human lineage after splitting from the other hominin branches, and most probably within the last 100,000 years (Chomsky, 2011 ; Crow, 1997 ; Klein & Edgar, 2002 ). Other accounts consider the evolutionary emergence of proficient spoken language as a multistage process involving multiple phenotypic components and multiple genomic changes (Fitch, 2012 ), with some hypotheses placing importance on gene–culture interactions (Fisher & Ridley, 2013 ; Laland, Odling-Smee, & Myles, 2010 ). Now that we have access to comprehensive descriptions of genomic changes along different branches of the hominin tree, it will become feasible to empirically evaluate different evolutionary accounts, to assess which are more compatible with the available molecular data.
For example, based on analyses of ancient genomes, it has been estimated that 96 amino-acid changes, in 87 protein-coding genes, have become fixed on the human lineage (that is, they are now shared by all humans in every population) after splitting from our common ancestor with Neandertals (Prüfer et al., 2014 ). Such comparisons also identified ~3,000 fixed changes, outside of protein-coding regions, with potential to impact on the regulation of gene expression, arising during this same period. Future research programmes can use bioinformatics, functional analyses in neuronal cell models, comparison to data from language disorders, and other screening methods, to systematically assess the likely functional impact of the various changes of interest, and their relevance to neural phenotypes. If it is assumed that Neandertals did not have language (a controversial issue as noted above) and that the unusual linguistic capacity of modern humans is the result of just a single relatively recent mutation of large effect (also subject to considerable debate), then it will very likely be contained within our existing catalogue of fixed human–Neandertal differences. Thus, although I am personally sceptical of such a single-mutation account, if this model is correct then the putative responsible mutation is entirely open to discovery via experimental means. To be clear, I do not doubt that sorting through the many plausible genomic changes is a difficult challenge, certainly at present when the available functional assays are still laborious and low-throughput. However, the key point is that this research programme (and other similar endeavours) is in principle a perfectly tractable one, well within the grasp of modern science. By paying attention to the relevant genomes themselves, we move away from unconstrained speculation about abstract genetic factors, to empirical evaluation of alternative perspectives on the origins of language.
It is important to recognize that language origins present us with an inverse problem; we will never precisely reconstruct the evolutionary history of our species. However, given the new sources of empirical data that can be brought to bear on the emergence of human traits, we are in a much stronger position to distinguish between the merits of different accounts of language evolution, and to generate novel hypotheses that are amenable to experimental testing.
- Ayub, Q., Yngvadottir, B., Chen, Y., Xue, Y., Hu, M., Vernes, S. C., …Tyler-Smith, C. (2013). FOXP2 targets show evidence of positive selection in European populations. American Journal of Human Genetics, 92, 696–706. doi:10.1016/j.ajhg.2013.03.019 [ PMC free article ] [ PubMed ]
- Bishop DVM. Genetic and environmental risks for specific language impairment in children. Philosophical Transactions of the Royal Society, B: Biological Sciences. 2001; 356 (1407):369–380. doi: 10.1098/rstb.2000.0770. [ PMC free article ] [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Bonkowsky JL, Chien CB. Molecular cloning and developmental expression of foxP2 in zebrafish. Developmental Dynamics. 2005; 234 (3):740–746. doi: 10.1002/dvdy.20504. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Crow TJ. Is schizophrenia the price that Homo sapiens pays for language? Schizophrenia Research. 1997; 28 :127–141. doi: 10.1016/S0920-9964(97)00110-2. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Chomsky N. Language and other cognitive systems. What is special about language? Language Learning and Development. 2011; 7 :263–278. doi: 10.1080/15475441.2011.584041. [ CrossRef ] [ Google Scholar ]
- Dediu D, Levinson SC. On the antiquity of language: The reinterpretation of Neandertal linguistic capacities and its consequences. Frontiers in Psychology. 2013; 4 :397. doi: 10.3389/fpsyg.2013.00397. [ PMC free article ] [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Deriziotis, P., O'Roak, B. J., Graham, S. A., Estruch, S. B., Dimitropoulou, D., Bernier, R. A., … Fisher, S. E. (2014). De novo TBR1 mutations in sporadic autism disrupt protein functions. Nature Communications , 5 , 4954. doi:10.1038/ncomms5954 [ PMC free article ] [ PubMed ]
- Enard W. FOXP2 and the role of cortico-basal ganglia circuits in speech and language evolution. Current Opinion in Neurobiology. 2011; 21 (3):415–424. doi: 10.1016/j.conb.2011.04.008. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Enard W. Mouse models of human evolution. Current Opinion in Genetics and Development. 2014; 29 :75–80. doi: 10.1016/j.gde.2014.08.008. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Enard, W., Przeworski, M., Fisher, S. E., Lai, C. S. L., Wiebe, V., Kitano, T., … Pääbo, S. (2002). Molecular evolution of FOXP2, a gene involved in speech and language. Nature, 418 (6900), 869–872 [ PubMed ]
- Fisher SE. Tangled webs: Tracing the connections between genes and cognition. Cognition. 2006; 101 :270–297. doi: 10.1016/j.cognition.2006.04.004. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Fisher SE, Marcus G. The eloquent ape: Genes, brains and the evolution of language. Nature Reviews. Genetics. 2006; 7 :9–20. doi: 10.1038/nrg1747. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Fisher SE, Ridley M. Culture, genes, and the human revolution. Science. 2013; 340 (6135):929–930. doi: 10.1126/science.1236171. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Fisher SE, Scharff C. FOXP2 as a molecular window into speech and language. Trends in Genetics. 2009; 25 :166–177. doi: 10.1016/j.tig.2009.03.002. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Fisher SE, Vernes SC. Genetics and the Language Sciences. Annual Review of Linguistics. 2015; 1 :289–310. doi: 10.1146/annurev-linguist-030514-125024. [ CrossRef ] [ Google Scholar ]
- Fitch WT. Evolutionary Developmental Biology and Human Language Evolution: Constraints on Adaptation. Evolutionary Biology. 2012; 39 (4):613–637. doi: 10.1007/s11692-012-9162-y. [ PMC free article ] [ PubMed ] [ CrossRef ] [ Google Scholar ]
- French CA, Jin X, Campbell TG, Gerfen E, Groszer M, Fisher SE, Costa RM. An aetiological Foxp2 mutation causes aberrant striatal activity and alters plasticity during skill learning. Molecular Psychiatry. 2012; 17 :1077–1085. doi: 10.1038/mp.2011.105. [ PMC free article ] [ PubMed ] [ CrossRef ] [ Google Scholar ]
- French CA, Fisher SE. What can mice tell us about Foxp2 function? Current Opinion in Neurobiology. 2014; 28 :72–79. doi: 10.1016/j.conb.2014.07.003. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Fu, Q., Li, H., Moorjani, P., Jay, F., Slepchenko, S. M., Bondarev, A. A., … Pääbo, S. (2014). Genome sequence of a 45,000-year-old modern human from western Siberia. Nature, 514 (7523), 445–449. doi:10.1038/nature13810 [ PMC free article ] [ PubMed ]
- Gialluisi, A., Newbury, D. F., Wilcutt, E. G., Olson, R. K., DeFries, J. C., Brandler, W. M., … Fisher, S. E. (2014). Genome-wide screening for DNA variants associated with reading and language traits. Genes, Brain and Behavior, 13, 686 – 701. doi:10.1111/gbb.12158 [ PMC free article ] [ PubMed ]
- Graham SA, Fisher SE. Understanding language from a genomic perspective. Annual Review of Genetics. 2015; 49 :131–160. doi: 10.1146/annurev-genet-120213-092236. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Graham SA, Deriziotis P, Fisher SE. Insights into the genetic foundations of human communication. Neuropsychology Review. 2015; 25 (1):3–26. doi: 10.1007/s11065-014-9277-2. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Groszer, M., Keays, D. A., Deacon, R. M. J., De Bono, J. P., Prasad-Mulcare, S., Gaub, S., … Fisher, S. E. (2008). Impaired synaptic plasticity and motor learning in mice with a point mutation implicated in human speech deficits. Current Biology, 18 (5), 354–362. doi:10.1016/j.cub.2008.01.060 [ PMC free article ] [ PubMed ]
- Haesler S, Rochefort C, Georgi B, Licznerski P, Osten P, Scharff C. Incomplete and inaccurate vocal imitation after knockdown of FoxP2 in songbird basal ganglia nucleus Area X. PLoS Biology. 2007; 5 (12):e321. doi: 10.1371/journal.pbio.0050321. [ PMC free article ] [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Haesler S, Wada K, Nshdejan A, Morrisey EE, Lints T, Jarvis ED, Scharff C. FoxP2 expression in avian vocal learners and non-learners. Journal of Neuroscience. 2004; 24 (13):3164–3175. doi: 10.1523/JNEUROSCI.4369-03.2004. [ PMC free article ] [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nature Reviews. Neuroscience. 2009; 10 :647–658. doi: 10.1038/nrn2699. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Kato M, Okanoya K, Koike T, Sasaki E, Okano H, Watanabe S, Iriki A. Human speech- and reading-related genes display partially overlapping expression patterns in the marmoset brain. Brain and Language. 2014; 133 :26–38. doi: 10.1016/j.bandl.2014.03.007. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Klein RG, Edgar B. The dawn of human culture. New York: Wiley; 2002. [ Google Scholar ]
- Kolodkin AL, Tessier-Lavigne M. Mechanisms and molecules of neuronal wiring: A primer. Cold Spring Harbor Perspectives in Biology. 2011; 3 (6):a001727. doi: 10.1101/cshperspect.a001727. [ PMC free article ] [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Krause, J., Lalueza-Fox, C., Orlando, L., Enard, W., Green, R. E., Burbano, H. A., … Pääbo, S. (2007). The derived FOXP2 variant of modern humans was shared with Neandertals. Current Biology, 17 (21), 1908 – 1912 [ PubMed ]
- Lai CSL, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001; 413 :519–523. doi: 10.1038/35097076. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Lai CSL, Gerrelli D, Monaco AP, Fisher SE, Copp AJ. FOXP2 expression during brain development coincides with adult sites of pathology in a severe speech and language disorder. Brain. 2003; 126 (11):2455–2462. doi: 10.1093/brain/awg247. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Laland KN, Odling-Smee J, Myles S. How culture shaped the human genome: Bringing genetics and the human sciences together. Nature Reviews. Genetics. 2010; 11 (2):137–148. doi: 10.1038/nrg2734. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Luciano, M., Evans, D. M., Hansell, N. K., Medland, S. E., Montgomery, G. W., Martin, N. G., … Bates, T. C. (2013). A genome-wide association study for reading and language abilities in two population cohorts. Genes, Brain and Behavior, 12 (6), 645–52. doi:10.1111/gbb.12053 [ PMC free article ] [ PubMed ]
- Maricic, T., Günther, V., Georgiev, O., Gehre, S., Curlin, M., Schreiweis, C., … Pääbo, S. (2013). A recent evolutionary change affects a regulatory element in the human FOXP2 gene. Molecular Biology and Evolution, 30 (4), 844 – 852. doi:10.1093/molbev/mss271 [ PubMed ]
- Meyer, M., Arsuaga, J. L., de Filippo, C., Nagel, S., Aximu-Petri, A., Nickel, B., … Pääbo, S. (2016). Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins. Nature, 531 (7595), 504–507. doi:10.1038/nature17405 [ PubMed ]
- Meyer, M., Kircher, M., Gansauge, M. T., Li, H., Racimo, F., Mallick, S., … Pääbo, S. (2012). A high-coverage genome sequence from an archaic Denisovan individual. Science, 338 (6104), 222–226. doi: 10.1126/science.1224344 [ PMC free article ] [ PubMed ]
- Mozzi, A., Forni, D., Clerici, M., Pozzoli, U., Mascheretti, S., Guerini, F. R., … Sironi, M. (2016). The evolutionary history of genes involved in spoken and written language: Beyond FOXP2. Scientific Reports, 6, 22157. doi:10.1038/srep22157 [ PMC free article ] [ PubMed ]
- Murugan M, Harward S, Scharff C, Mooney R. Diminished FoxP2 levels affect dopaminergic modulation of corticostriatal signaling important to song variability. Neuron. 2013; 80 (6):1464–1476. doi: 10.1016/j.neuron.2013.09.021. [ PMC free article ] [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Narasimhan, V. M., Hunt, K. A., Mason, D., Baker, C. L., Karczewski, K. J., Barnes, M. R., … van Heel, D. A. (2016). Health and population effects of rare gene knockouts in adult humans with related parents. Science, 352 (6284), 474 – 477. doi:10.1126/science.aac8624 [ PMC free article ] [ PubMed ]
- Newbury DF, Monaco AP. Genetic advances in the study of speech and language disorders. Neuron. 2010; 68 (2):309–320. doi: 10.1016/j.neuron.2010.10.001. [ PMC free article ] [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Pääbo S. The human condition-a molecular approach. Cell. 2014; 157 (1):216–226. doi: 10.1016/j.cell.2013.12.036. [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Prüfer, K., Racimo, F., Patterson, N., Jay, F., Sankararaman, S., Sawyer, S., … Pääbo, S. (2014). The complete genome sequence of a Neanderthal from the Altai Mountains. Nature, 505 (7481), 43–49. doi:10.1038/nature12886 [ PMC free article ] [ PubMed ]
- Scharff C, Petri J. Evo-devo, deep homology and FoxP2: Implications for the evolution of speech and language. Philosophical Transactions of the Royal Society, B: Biological Sciences. 2011; 366 (1574):2124–2140. doi: 10.1098/rstb.2011.0001. [ PMC free article ] [ PubMed ] [ CrossRef ] [ Google Scholar ]
- St Pourcain, B., Cents, R. A. M., Whitehouse, A. J. O., Haworth, C. M. A., Davis, O. S. P., O’Reilly, P. F., … Davey Smith, G. (2014). Common variation near ROBO2 is associated with expressive vocabulary in infancy. Nature Communications, 5, 4831, doi:10.1038/ncomms5831 [ PMC free article ] [ PubMed ]
- Sollis, E., Graham, S. A., Vino, A., Froehlich, H., Vreeburg, M., Dimitropoulou, D., … Fisher, S. E. (2016). Identification and functional characterization of de novo FOXP1 variants provides novel insights into the etiology of neurodevelopmental disorder. Human Molecular Genetics, 25 (3), 546–557. doi:10.1093/hmg/ddv495 [ PubMed ]
- Tan X, Shi SH. Neocortical neurogenesis and neuronal migration. Wiley Interdisciplinary Reviews: Developmental Biology. 2013; 2 :443–459. doi: 10.1002/wdev.88. [ PMC free article ] [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Teramitsu I, Kudo LC, London SE, Geschwind DH, White SA. Parallel FoxP1 and FoxP2 expression in songbird and human brain predicts functional interaction. Journal of Neuroscience. 2004; 24 (13):3152–3163. doi: 10.1523/JNEUROSCI.5589-03.2004. [ PMC free article ] [ PubMed ] [ CrossRef ] [ Google Scholar ]
- The 1000 Genomes Project Consortium A global reference for human genetic variation. Nature. 2015; 526 (7571):68–74. doi: 10.1038/nature15393. [ PMC free article ] [ PubMed ] [ CrossRef ] [ Google Scholar ]
- Vernes, S. C., Newbury, D. F., Abrahams, B. S., Winchester, L., Nicod, J., Groszer, M., … Fisher, S. E. (2008). A functional genetic link between distinct developmental language disorders. New England Journal of Medicine, 359 (22), 2337–2345. doi:10.1056/NEJMoa0802828 [ PMC free article ] [ PubMed ]
- Vernes, S. C., Spiteri, E., Nicod, J., Groszer, M., Taylor, J. M., Davies, K. E., … Fisher, S. E. (2007). High-throughput analysis of promoter occupancy reveals direct neural targets of FOXP2, a gene mutated in speech and language disorders. American Journal of Human Genetics, 81 (6), 1232–1250. doi:10.1086/522238 [ PMC free article ] [ PubMed ]
- Villanueva, P., Nudel, R., Hoischen, A., Fernández, M. A., Simpson, N. H., Gilissen, C., … Newbury, D. F. (2015). Exome sequencing in an admixed isolated population indicates NFXL1 variants confer a risk for Specific Language Impairment. PLoS Genetics, 11 (3), e1004925. doi:10.1371/journal.pgen.1004925 [ PMC free article ] [ PubMed ]
- Wohlgemuth S, Adam I, Scharff C. FoxP2 in songbirds. Current Opinion in Neurobiology. 2014; 28 :86–93. doi: 10.1016/j.conb.2014.06.009. [ PubMed ] [ CrossRef ] [ Google Scholar ]
Communication Sciences and Disorders
Us news & world report ranks iowa audiology, speech-language pathology among the top in the nation.
Two programs in the Department of Communication Sciences and Disorders are once again recognized among the best in their field according to the U.S. News & World Report Best Graduate Schools rankings for 2024 .
Iowa’s audiology program is again ranked second best in the nation and remains the top public institution training audiologists. Speech-Language Pathology climbed in the rankings to fifth and is now the third-ranked public institution.
New rankings of nation-wide Audiology and Speech-Language Pathology graduate programs is available on the USNWR website.
“These rankings recognize the daily efforts of our remarkable faculty and staff, who consistently strive to provide exceptional education to our students,” said Eric Hunter , DEO and Harriet B. and Harold S. Brady Chair in Liberal Arts and Sciences. “We are focused on training outstanding audiologists and speech-language pathologists.”
“Together, we are shaping the future of audiology and speech-language pathology, and we will continue to lead the way in delivering excellence,” Hunter added.
Hunter, who has been at Iowa since August 2023 and is a nationally recognized expert in the field, was the first hire under the University of Iowa's Transformational Faculty Hiring Program , which is aimed at attracting world-class faculty to strategic programs and areas of excellence. He received a PhD from Iowa’s Department of Communication Sciences and Disorders program in 2001.
Iowa has long been a leader in communication sciences and disorders dating back to 1897, when the university, led by Carl Seashore’s pioneering work, developed speech pathology as a discipline of study.
“CLAS is proud of our faculty and staff who continue to guide this storied program into the future,” said Dean Sara Sanders. “Because of their tremendous talent and dedication to their research and teaching, CSD continues to be at the forefront of audiology and speech-language pathology.”
The university, college, and department continue to lead, ensuring students access to unparalleled opportunities. Construction has started on a new $249 million building that will provide a state-of-the-art learning space for Iowa students studying in the Department of Communication Sciences and Disorders, Department of Health and Human Physiology, and the Carver College of Medicine’s Department of Physical Therapy and Rehabilitation Science.
Substantial completion of the building is anticipated in 2025.
NOTICE: The University of Iowa Center for Advancement is an operational name for the State University of Iowa Foundation, an independent, Iowa nonprofit corporation organized as a 501(c)(3) tax-exempt, publicly supported charitable entity working to advance the University of Iowa. Please review its full disclosure statement.
[Molecular genetic analysis of TUB18 and TUB20 intragenic polymorphism and various mutations of the CFTR gene in the Moscow region]
- PMID: 9445824
Allelic frequencies of two intron polymorphisms in the cystic fibrosis transmembrane regulator (CFTR) gene, TUB18 and TUB20, were estimated on chromosomes of 67 cystic fibrosis patients and on that of 37 healthy donors from Moscow and the Moscow oblast. Allele 2 of the TUB 18, and allele 1 of the TUB20 were 2.1 and 1.5 times more frequent on the non-delta F508 chromosomes of the cystic fibrosis patients than on chromosomes of healthy donors, i.e. these alleles were in linkage disequilibrium with the CFTR gene. Allele 1 of the TUB18 marker and allele 2 of the TUB20 marker demonstrated absolute linkage disequilibrium with the delta F508 mutation of the CFTR gene. The degree of association between the TUB18 and TUB20 intron polymorphisms and the GATT and T854T intragenic polymorphisms was analyzed. Of all 62 delta F508 chromosomes tested, 98.3% shared the 2-1-1-2 GATT- T854T-TUB18-TUB20 haplotype. Eight major (more frequent) GATT-T854T-TUB18-TUB20 haplotypes were found in 89.5% of normal, and in 97.9% of non-delta F508 chromosomes of cystic fibrosis patients from the Moscow region. Three of these major haplotypes, 2-1-1-2, 1-2-2-1, and 2-2-1-2, were respectively 2.5, 2, and 1.5 times more frequent on non-delta F508 cystic fibrosis chromosomes than on normal chromosomes. Data on screening for the G542X, N1303K, and 394delTT mutations of the CFTR gene, carried out on 134 chromosomes of cystic fibrosis patients from the Moscow region are presented. The frequencies of the G542X and 394delTT mutations were estimated as 1.5%, while the frequency of the N1303K mutation was 2.2%.
Publication types
- English Abstract
- Case-Control Studies
- Cystic Fibrosis / genetics*
- Gene Frequency
- Genetic Markers*
- Point Mutation*
- Polymorphism, Genetic*
- Genetic Markers
IMAGES
VIDEO
COMMENTS
The discovery of FOXP2 as the first gene implicated in speech and language disorders provided a window into the biology of speech and language. ... There are three areas which may be particularly fruitful in unravelling the biology of language disorders. Gene-gene interaction, also known as epistasis, is when two independent variants interact ...
FOXP2-related speech and language disorder (FOXP2-SLD) is caused by heterozygous FOXP2 pathogenic variants (including whole- or partial-gene deletions). The core phenotype of FOXP2-SLD is childhood apraxia of speech (CAS), a disorder of speech motor programming or planning that affects the production, sequencing, timing, and stress of sounds, and the accurate sequencing of speech sounds into ...
Speech control. Children with Foxp2-associated apraxia tend to begin speaking later than other children, and their speech is often difficult to understand. The disorder is believed to arise from impairments in brain regions, such as the striatum, that control the movements of the lips, mouth, and tongue.
If there is mention of speech or language impairment, ... Coleman M, Braden RO, et al. Severe childhood speech disorder: Gene discovery highlights transcriptional dysregulation. Neurology 2020;94 ...
In 2001, investigation of a large three generational family with severe speech disorder, known as childhood apraxia of speech (CAS), revealed the first causative gene; FOXP2. A long hiatus then ...
It is the first gene to be linked to an inherited form of language and speech disorder. 4 The discovery of a mutation in FOXP2 in a family with a speech and language disorder opens a new window to understanding of the genetic cascades and neural circuits that underlie speech and language via molecular approaches.
Moreover, speech and other communication phenotypes follow a developmental trajectory, where some speech and language disorders resolve with age, whereas others persist; genetic influences on the ...
Rare genetic variants that disrupt speech development provide entry points for deciphering the neurobiological foundations of key human capacities. The value of this approach is illustrated by FOXP2, a transcription factor gene that was implicated in speech apraxia, and subsequently investigated using human cell-based systems and animal models.
One notable success in this area was the discovery that heterozygous disruptions of the FOXP2 gene cause a rare mendelian speech and language disorder. 5-9 Point mutations and chromosomal ...
The discovery of FOXP2 as the first gene implicated in speech and language disorders provided a window into the biology of speech and language. FOXP2, a transcription factor, is involved in the downstream control of many other genes important for a huge range of biological processes.
Speech apraxia. The first gene to be clearly implicated in a speech and language disorder was FOXP2.Disruptions of this gene cause a monogenic form of developmental verbal dyspraxia (DVD), also known as childhood apraxia of speech (CAS) [], characterized by problems with the learning and execution of coordinated movement sequences of the mouth, tongue, lips and soft palate [18, 19].
Over a decade and a half ago, investigation of a large multigenerational family and an independent translocation case uncovered the first mutations to be implicated in a monogenic speech and language disorder [2].Since then, the FOXP2 transcription factor gene has provided a paradigm for bridging genes, neurons, brains, and spoken language [5]. ...
As noted above, the vast majority of cases of language impairment are likely to have a complex genetic basis. However, in the late 1980s clinical geneticists came across an unusual large family showing an apparently simple inheritance pattern for their speech and language problems [].In this pedigree, known as the 'KE family', approximately half of the 30 family members, spread over three ...
Vocal communication mediated by speech and language is a uniquely human trait, and has served an important evolutionary role in the development of our species. Deficits in speech and language functions can be of numerous types, including aphasia, stuttering, articulation disorders, verbal dyspraxia, and specific language impairment; language ...
Language Impairment (LI): A developmental language disorder that can affect both expressive and receptive language and impairs the ability to understand and/or use words in context. The estimated prevalence of LI at kindergarten is 7.4%.7. Reading Disorders (RD): A learning disorder that involves signifi-cant impairment of reading accuracy ...
Our findings show that poor communication is a central feature of SETBP1 haploinsufficiency disorder, confirming this gene as a strong candidate for speech and language disorders. You have full ...
The estimated prevalence of speech and language disorders ranges between 3% and 16% of U.S. children and adolescents aged 3 to 21 years. Boys are more than twice as likely to be affected than girls.
speech sound disorder (American Speech-Language-Hearing Association [ASHA], 2007). Although the evidence for CAS in some autistic indi-viduals is growing (Chenausky et al., 2019; Chenausky, Baas, et al., 2023; Tierney et al., 2015; Vashdi et al., 2020), our comprehensive review of the literature suggests the ...
The first gene to be implicated in a speech and language disorder was identified by the investigation of a large family affected by a distinctive form of speech impairment known as verbal dyspraxia. Verbal dyspraxia is characterized by difficulties in the control of orofacial muscles leading to a deficit in the production of fluent speech.
The first gene to be implicated in a speech and language disorder was identified by the investigation of a large family affected by a distinctive form of speech impairment known as verbal dyspraxia. Verbal dyspraxia is characterized by difficulties in the control of orofacial muscles leading to a deficit in the production of fluent speech.
Purpose:The word learning of preschool-age children with developmental language disorder (DLD) is improved when spaced retrieval practice is incorporated into the learning sessions. ... The challenge of rich vocabulary instruction for children with developmental language disorder. Language, Speech, and Hearing Services in Schools, 52(2), 467 ...
The role of foreign accent and short -term exposure on speech in speech recognition" Talk B4 Charlotte Poulisse 1, Katrien Segaert , Ali Mazaheri , Linda Wheeldon2; 1University of Birmingham, 2University of Agder "The oscillatory mechanisms supporting syntactic language comprehension in healthy aging" Coffee break Talk C1 , 4
A decade of disproportionality: A state-level analysis of African American students enrolled in the primary disability category of speech or language impairment. Language, Speech, and Hearing Services in Schools , 50(2), 267-282.
An additional PstI fragment of 197 bp was found to be present in intron 1 of the gene encoding chicken growth hormone which we reported previously [Tanaka et al., Gene 112(1992)235-239]. Read more
Scientists believe gene regulation, one of life's most basic functions, holds the key to unlocking the origins of disease and disorder. Language models like this one could provide a new way to probe. Wang's collaborators include researchers from the biotech firm RVAC Medicines as well as the Stanford University School of Medicine.
Using electrophoretic mobility shift assay the proteins, specifically bound to the double-stranded oligonucleotides corresponding the binding sites of the transcription factors of AP1 family, NF-IL6 and SP1 involved in the up-regulation of human multidrug resistance (mdr1) gene and also proteins bound to the oligonucleotide corresponding to the element responsive for the stimulation by serum ...
Having pinpointed a gene involved in a language-related disorder, purely based on genomic data from families and cases, ... In addition, the mutations that yield speech and language impairments can be directly introduced into cells grown in the laboratory, or into animal models, to help understand how the crucial mechanisms and pathways go awry ...
The speech-language pathology program climbed in the rankings to fifth and is now the third-ranked public institution. CSD is housed in the College of Liberal Arts and Sciences. ... Iowa has long been a leader in communication sciences and disorders dating back to 1897, when the university, led by Carl Seashore's pioneering work, developed ...
In addition to motor symptoms, neurocognitive impairment (NCI) affects patients with prodromal Parkinson's disease (PD). NCI in PD ranges from subjective cognitive complaints to dementia. The purpose of this review is to present the available evidence of NCI in PD and highlight the heterogeneity of NCI phenotypes as well as the range of factors that contribute to NCI onset and progression. A ...
Allelic frequencies of two intron polymorphisms in the cystic fibrosis transmembrane regulator (CFTR) gene, TUB18 and TUB20, were estimated on chromosomes of 67 cystic fibrosis patients and on that of 37 healthy donors from Moscow and the Moscow oblast. Allele 2 of the TUB 18, and allele 1 of the TUB20 were 2.1 and 1.5 times more frequent on ...