Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- News & Views
- Published: 10 February 2022
GENE THERAPY

A solid start for gene therapy in Tay–Sachs disease
- Timothy W. Yu ORCID: orcid.org/0000-0003-2988-7701 1 , 2 , 3 , 4 &
- Olaf Bodamer 1 , 2 , 3
Nature Medicine volume 28 , pages 236–237 ( 2022 ) Cite this article
2925 Accesses
2 Citations
23 Altmetric
Metrics details
- Drug development
- Paediatric neurological disorders
- Translational research
A ‘gene addition’ strategy shows promise in a first-in-human trial involving two infant patients.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
24,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
195,33 € per year
only 16,28 € per issue
Rent or buy this article
Prices vary by article type
Prices may be subject to local taxes which are calculated during checkout
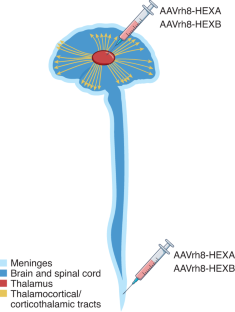
Flotte, T. R. et al. Nat. Med. https://doi.org/10.1038/s41591-021-01664-4 (2022).
Article PubMed Google Scholar
Platt, F. M., d’Azzo, A., Davidson, B. L., Neufeld, E. F. & Tifft, C. J. Nat. Rev. Dis. Primers 4 , 274 (2018).
Google Scholar
Bembi, B. et al. Neurology 66 , 278–280 (2006).
Article CAS Google Scholar
Maegawa, G. H. B. et al. Mol. Genet. Metab. 98 , 215–224 (2009).
Hwu, W. L. et al. Sci. Transl. Med. 4 , 134ra61 (2012).
Article Google Scholar
Mendell, J. R. et al. N. Engl. J. Med. 377 , 1713–1722 (2017).
Bailey, R. M., Armao, D., Nagabhushan Kalburgi, S. & Gray, S. J. Mol. Ther. Methods Clin. Dev. 9 , 160–171 (2018).
Bainbridge, J. W. B. et al. N. Engl. J. Med. 372 , 1887–1897 (2015).
Eichler, F. et al. N. Engl. J. Med. 377 , 1630–1638 (2017).
Download references
Author information
Authors and affiliations.
Division of Genetics and Genomics, Boston Children’s Hospital, Boston, MA, USA
Timothy W. Yu & Olaf Bodamer
Broad Institute of MIT and Harvard, Cambridge, MA, USA
Harvard Medical School, Boston, MA, USA
Department of Neurology, Boston Children’s Hospital, Boston, MA, USA
Timothy W. Yu
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Timothy W. Yu .
Ethics declarations
Competing interests.
The authors declare no competing interests.
Rights and permissions
Reprints and permissions
About this article
Cite this article.
Yu, T.W., Bodamer, O. A solid start for gene therapy in Tay–Sachs disease. Nat Med 28 , 236–237 (2022). https://doi.org/10.1038/s41591-022-01687-5
Download citation
Published : 10 February 2022
Issue Date : February 2022
DOI : https://doi.org/10.1038/s41591-022-01687-5
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing: Translational Research newsletter — top stories in biotechnology, drug discovery and pharma.

- Download PDF
- Share X Facebook Email LinkedIn
- Permissions
Tay-Sachs Disease
Author Affiliations : University Hospitals of Cleveland, Case Western Reserve University School of Medicine, Cleveland, Ohio.
CHRISTOPHER G.GOETZMD
Tay-Sachs disease is an autosomal recessive disease caused by a deficiency of β-hexosaminidase A, the lysosomal enzyme that normally degrades GM2 ganglioside. As a result, GM2 ganglioside accumulates in the lysosomes of nerve cells. The disease is one of a family of lysosomal storage disorders known as GM2 gangliosidoses, each determined by the specific peptide (α and β subunits of β-hexosaminidase A and the GM2 activator protein) that is defective in the degradation of GM2 ganglioside. 1 While Tay-Sachs disease commonly refers to the classic infantile form of this GM2 gangliosidosis (also called type 1 GM2 gangliosidosis), wherein β-hexosaminidase A is virtually absent, juvenile and late-onset forms also occur when there is residual enzymatic activity. The highest carrier rate has been among Ashkenazic Jews, although the incidence has decreased among this population because of widespread carrier screening, while clusters remain among certain French Canadian and Cajun populations. This article in the Seminal Citations series focuses on early descriptions of the disease and key developments in biochemistry, genetics, testing, and treatment.
Read More About
Fernandes Filho JA , Shapiro BE. Tay-Sachs Disease. Arch Neurol. 2004;61(9):1466–1468. doi:10.1001/archneur.61.9.1466
Manage citations:
© 2024
Artificial Intelligence Resource Center
Neurology in JAMA : Read the Latest
Browse and subscribe to JAMA Network podcasts!
Others Also Liked
Select your interests.
Customize your JAMA Network experience by selecting one or more topics from the list below.
- Academic Medicine
- Acid Base, Electrolytes, Fluids
- Allergy and Clinical Immunology
- American Indian or Alaska Natives
- Anesthesiology
- Anticoagulation
- Art and Images in Psychiatry
- Artificial Intelligence
- Assisted Reproduction
- Bleeding and Transfusion
- Caring for the Critically Ill Patient
- Challenges in Clinical Electrocardiography
- Climate and Health
- Climate Change
- Clinical Challenge
- Clinical Decision Support
- Clinical Implications of Basic Neuroscience
- Clinical Pharmacy and Pharmacology
- Complementary and Alternative Medicine
- Consensus Statements
- Coronavirus (COVID-19)
- Critical Care Medicine
- Cultural Competency
- Dental Medicine
- Dermatology
- Diabetes and Endocrinology
- Diagnostic Test Interpretation
- Drug Development
- Electronic Health Records
- Emergency Medicine
- End of Life, Hospice, Palliative Care
- Environmental Health
- Equity, Diversity, and Inclusion
- Facial Plastic Surgery
- Gastroenterology and Hepatology
- Genetics and Genomics
- Genomics and Precision Health
- Global Health
- Guide to Statistics and Methods
- Hair Disorders
- Health Care Delivery Models
- Health Care Economics, Insurance, Payment
- Health Care Quality
- Health Care Reform
- Health Care Safety
- Health Care Workforce
- Health Disparities
- Health Inequities
- Health Policy
- Health Systems Science
- History of Medicine
- Hypertension
- Images in Neurology
- Implementation Science
- Infectious Diseases
- Innovations in Health Care Delivery
- JAMA Infographic
- Law and Medicine
- Leading Change
- Less is More
- LGBTQIA Medicine
- Lifestyle Behaviors
- Medical Coding
- Medical Devices and Equipment
- Medical Education
- Medical Education and Training
- Medical Journals and Publishing
- Mobile Health and Telemedicine
- Narrative Medicine
- Neuroscience and Psychiatry
- Notable Notes
- Nutrition, Obesity, Exercise
- Obstetrics and Gynecology
- Occupational Health
- Ophthalmology
- Orthopedics
- Otolaryngology
- Pain Medicine
- Palliative Care
- Pathology and Laboratory Medicine
- Patient Care
- Patient Information
- Performance Improvement
- Performance Measures
- Perioperative Care and Consultation
- Pharmacoeconomics
- Pharmacoepidemiology
- Pharmacogenetics
- Pharmacy and Clinical Pharmacology
- Physical Medicine and Rehabilitation
- Physical Therapy
- Physician Leadership
- Population Health
- Primary Care
- Professional Well-being
- Professionalism
- Psychiatry and Behavioral Health
- Public Health
- Pulmonary Medicine
- Regulatory Agencies
- Reproductive Health
- Research, Methods, Statistics
- Resuscitation
- Rheumatology
- Risk Management
- Scientific Discovery and the Future of Medicine
- Shared Decision Making and Communication
- Sleep Medicine
- Sports Medicine
- Stem Cell Transplantation
- Substance Use and Addiction Medicine
- Surgical Innovation
- Surgical Pearls
- Teachable Moment
- Technology and Finance
- The Art of JAMA
- The Arts and Medicine
- The Rational Clinical Examination
- Tobacco and e-Cigarettes
- Translational Medicine
- Trauma and Injury
- Treatment Adherence
- Ultrasonography
- Users' Guide to the Medical Literature
- Vaccination
- Venous Thromboembolism
- Veterans Health
- Women's Health
- Workflow and Process
- Wound Care, Infection, Healing
- Register for email alerts with links to free full-text articles
- Access PDFs of free articles
- Manage your interests
- Save searches and receive search alerts
- Introduction to Genomics
- Educational Resources
- Policy Issues in Genomics
- The Human Genome Project
- Funding Opportunities
- Funded Programs & Projects
- Division and Program Directors
- Scientific Program Analysts
- Contact by Research Area
- News & Events
- Research Areas
- Research investigators
- Research Projects
- Clinical Research
- Data Tools & Resources
- Genomics & Medicine
- Family Health History
- For Patients & Families
- For Health Professionals
- Jobs at NHGRI
- Training at NHGRI
- Funding for Research Training
- Professional Development Programs
- NHGRI Culture
- Social Media
- Broadcast Media
- Image Gallery
- Press Resources
- Organization
- NHGRI Director
- Mission & Vision
- Policies & Guidance
- Institute Advisors
- Strategic Vision
- Leadership Initiatives
- Diversity, Equity, and Inclusion
- Partner with NHGRI
- Staff Search
About Tay-Sachs Disease
Tay-Sachs disease is a fatal genetic disorder that results in progressive destruction of the nervous system.
What Do We Know About Heredity and Tay-Sachs Disease?
Tay-Sachs disease (TSD) is a fatal genetic disorder, most commonly occurring in children, that results in progressive destruction of the nervous system. Tay-Sachs is caused by the absence of a vital enzyme called hexosaminidase-A (Hex-A). Without Hex-A, a fatty substance, or lipid, called GM2 ganglioside accumulates abnormally in cells, especially in the nerve cells of the brain. This ongoing accumulation causes progressive damage to the cells.
In children, the destructive process begins in the fetus early in pregnancy. However, a baby with Tay-Sachs disease appears normal until about six months of age when its development slows. By about two years of age, most children experience recurrent seizures and diminishing mental function. The infant gradually regresses, and is eventually unable to crawl, turn over, sit or reach out. Eventually, the child becomes blind, cognitively impaired, paralyzed and non-responsive. By the time a child with Tay-Sachs is three or four years old, the nervous system is so badly affected that death usually results by age five.
A much rarer form of Tay-Sachs, Late-Onset Tay-Sachs disease, affects adults and causes neurological and intellectual impairment. Only recently identified, the disease has not been extensively described. As for the childhood form of Tay-Sachs, there is no cure. Treatment involves managing the symptoms of the disease.
Defect in Hex-A Gene Causes Tay-Sachs:
Tay-Sachs disease results from defects in a gene on chromosome 15 that codes for production of the enzyme Hex-A. We all have two copies of this gene. If either or both Hex-A genes are active, the body produces enough of the enzyme to prevent the abnormal build-up of the GM2 ganglioside lipid. Carriers of Tay-Sachs - people who have one copy of the inactive gene along with one copy of the active gene - are healthy. They do not have Tay-Sachs disease but they may pass on the faulty gene to their children.
Carriers have a 50 percent chance of passing on the defective gene to their children. A child who inherits one inactive gene is a Tay-Sachs carrier like the parent. If both parents are carriers and their child inherits the defective Hex-A gene from each of them, the child will have Tay-Sachs disease. When both parents are carriers of the defective Tay-Sachs gene, each child has a 25 percent chance of having Tay-Sachs disease and a 50 percent chance of being a carrier.
Eastern European (Ashkenazi) Jews at Greater Risk for Tay-Sachs Disease:
While anyone can be a carrier of Tay-Sachs, the incidence of the disease is significantly higher among people of eastern European (Ashkenazi) Jewish descent. Approximately one in every 27 Jews in the United States is a carrier of the Tay-Sachs disease gene. Non-Jewish French Canadians living near the St. Lawrence River and in the Cajun community of Louisiana also have a higher incidence of Tay-Sachs. For the general population, about one in 250 people are carriers.
There is no cure or effective treatment for Tay-Sachs disease. However, researchers are pursuing several approaches to finding a cure. Scientists are exploring enzyme replacement therapy to provide the Hex-A that is lacking in babies with Tay-Sachs. Bone marrow transplantation has been attempted also, but to date has not been successful in reversing or slowing damage to the central nervous system in babies with Tay-Sachs. Another avenue of research is gene therapy in which scientists transfer a normal gene into cells to replace an abnormal gene. This approach holds great promise for future Tay-Sachs patients.
Is there a test for Tay-Sachs disease?
A simple blood test can identify Tay-Sachs carriers. Blood samples can be analyzed by either enzyme assay or DNA studies. The enzyme assay is a biochemical test that measures the level of Hex-A in a person's blood. Carriers have less Hex-A in their body fluid and cells than non-carriers.
DNA-based carrier testing looks for specific mutations or changes in the gene that codes for Hex-A. Since 1985, when the Hex-A gene was isolated, more than 50 different mutations in this gene have been identified. Nevertheless, some mutations are not yet known. The current tests detect about 95 percent of carriers of Ashkenazi Jewish background and about 60 percent of carriers in the general population.
If both parents are carriers, they may want to consult with a genetic counselor for help in deciding whether to conceive or whether to have a fetus tested for Tay-Sachs. Extensive carrier testing of Ashkenazi Jews has significantly reduced the number of Tay-Sachs children in this population group. Today most cases of Tay-Sachs disease occur in populations thought not to be at high risk.
Prenatal testing for Tay-Sachs can be performed around the 11th week of pregnancy using chorionic villi sampling (CVS). This involves removing a tiny piece of the placenta. Alternatively, the fetus can be tested with amniocentesis around the 16th week of pregnancy. In this procedure, a needle is used to remove and test a sample of the fluid surrounding the baby.
Assisted reproductive therapy is an option for carrier couples who don't want to risk giving birth to a child with Tay-Sachs. This new technique used in conjunction with in-vitro fertilization enables parents who are Tay-Sachs carriers to give birth to healthy babies. Embryos created in-vitro are tested for Tay-Sachs genetic mutations before being implanted into the mother, allowing only healthy embryos to be selected.
Additional Resources
Medline Plus: Tay-Sachs Disease
NINDS: Tay-Sachs Disease Information Page
Genetics Home Reference: Tay-Sachs disease
GARD: Tay-Sachs Disease
Last updated: March 17, 2011
Gene Replacement Therapy
The National Institutes of Health (NIH) recently awarded the Tay-Sachs Gene Therapy Consortium (Consortium) a four-year, $3.5 million grant to continue its cutting-edge research to cure this fatal disease. The Consortium, formed in March 2007, is an international collaborative group of scientists from four institutions: Auburn University, Boston College, Cambridge University (U.K), and the Massachusetts General Hospital/Harvard University.
Children born with Tay-Sachs disease lack a vital enzyme, this substance accumulates abnormally and causes progressive damage until the nervous system can no longer sustain life. Researchers in the Consortium have refined a viral vector carrying two human genes- Hex-A, which is deficient in Tay-Sachs disease, and Hex-B, which is deficient in Sandhoff disease. They have successfully injected these genes into the brains of laboratory mice, prompting production of the missing enzyme at levels sufficient to correct the deficiency. However, curing affected mice is just one very promising step in their research.
The next task for the Consortium is to translate the results into advances in a large animal model of the disease, specifically cats. A cat’s brain is seen as an intermediary between mouse and human in terms of size and complexity. Al of this will hopefully translate into the ultimate goal of the Consortium- to develop a clinical trial of the gene therapy safe enough for humans within the next four years. If successful, it could potentially stop the progression of Tay-Sachs disease, although it would not be able to undo damage already done to the affected individuals.
For more information about this research, please visit the Tay-Sachs Gene Therapy Consortium’s website at www.tsgtconsortium.com .
Cord Blood Transplants
Cord blood transplants to treat genetic metabolic disorders in babies while they are still in the womb is a method which uses a small select number of therapeutic stem cells injected directly into the fetus’s abdomen at 12-14 weeks pregnancy. The idea is to give the baby cord blood stem cells from a healthy donor with the potential to provide healthy genes that will hopefully replace the genes that aren’t working in the baby’s cells.
There is a pilot trial currently open to pregnant women at risk for having children with fatal metabolic disorders including Tay-Sachs and Sandhoff diseases. The trial is being conducted out of Duke University Medical Center in Durham, N.C.
For a more complete listing of current research projects, please visit www.ntsad.org or www.curetay-sachs.org .
What is a Rare Disease?
A rare disease is defined as a condition that affects fewer than 1 in 200,000 patients in the United States or 1 in 2000 in Europe.
Many rare diseases are genetic (caused by change in DNA), which change can be inherited, spontaneous, or epigenetic. Since there are many genes (~20,000), there are many possible defects.
To date, about 7000 Rare Diseases have been identified.
Tay-Sachs disease
Disease researchers.
Specialists who have done research into Tay-Sachs disease.
These specialists have recieved grants, written articles, run clinical trials, or taken part in organizations relating to Tay-Sachs disease, and are considered knowledgeable about the disease as a result.
Clinical Trials
A clinical trial is how pharmaceutical companies and the FDA determine if treatment for a rare disease is safe and effective. Because the number of patients with rare diseases are extremely small, it is difficult for the companies to enroll enough patients to statistically prove (that the improvement wasn't just by chance) that the treatment was effective. It may take many years to treat enough patients to determine if a treatment is effective. The FDA, patient communities, legislation, and the drug companies are working on ways to address this issue.
Source: ClinicalTrials.gov
The National Institutes of Health sponsors a large number of grants each year in different areas of public health interest. As with all government spending, the funds allocated to each researcher, their research topic, and their results are available for review. InfoHub collects that data so you can see who is doing research in a particular area of rare diseases and what progress has been made.
FDA Orphan Drugs
Orphan Drug is a special status given by the FDA to a medication that was specifically developed for treatment of a rare disease. This status provides pharmaceutical companies with financial incentives for developing and marketing the drug.
Patient assistance programs can help patients pay for expensive prescriptions. There are often eligibility restrictions based on income, insurance status, and citizenship.
Before taking any medication, always check with a qualified professional for healthcare information, treatment advice.
Source: FDA Orphan Drug Database
FDA Orange Book Drugs
The FDA Orange Book drug database contains all approved drugs. Some drugs may be re-purposed from their original use to treat a rare disease. This table lists research papers that mention a drug in conjuntion with this disease.
Source: FDA Approved Drug Products
Chemotext is a publicly-available Webserver that mines the published literature in PubMed in the form of Medical Subject Headings (MeSH) terms.
The goal of Chemotext is to enable text-based drug-target-disease relationships in order to identify novel drug repurposing candidates and discovery targets.
Use this link to query the Chemotext database for Tay-Sachs disease
Use this link to visit the Chemotext homepage
- Patient Care & Health Information
- Diseases & Conditions
- Tay-Sachs disease
Tay-Sachs disease is a rare genetic disorder passed from parents to child. It's caused by the absence of an enzyme that helps break down fatty substances. These fatty substances, called gangliosides, build up to toxic levels in the brain and spinal cord and affect the function of the nerve cells.
In the most common and severe form of Tay-Sachs disease, signs and symptoms start to show up at about 3 to 6 months of age. As the disease progresses, development slows and muscles begin to weaken. Over time, this leads to seizures, vision and hearing loss, paralysis, and other major issues. Children with this form of Tay-Sachs disease typically live only a few years.
Less commonly, some children have the juvenile form of Tay-Sachs disease and may live into their teen years. Rarely, some adults have a late-onset form of Tay-Sachs disease which is often less severe than forms that begin in childhood.
If you have a family history of Tay-Sachs disease or if you're a member of a high-risk group and plan to have children, health care providers strongly recommend genetic testing and genetic counseling.
Products & Services
- A Book: Mayo Clinic Family Health Book, 5th Edition
- Newsletter: Mayo Clinic Health Letter — Digital Edition
There are three forms of Tay-Sachs disease: infantile, juvenile and late onset/adult.
Infantile form
In the most common and severe form, called infantile form, an infant typically begins showing signs and symptoms by about 3 to 6 months of age. Survival is usually only a few years. Signs and symptoms can include:
- Exaggerated startle response when the baby hears loud noises
- "Cherry-red" spots in the eyes
- Loss of motor skills, including turning over, crawling and sitting up
- Muscle weakness, progressing to paralysis
- Movement problems
- Vision loss and blindness
- Hearing loss and deafness
- Problems swallowing
- Loss of mental functions and a lack of response to surroundings
- Growth in head size (progressive macrocephaly)
Juvenile form
The juvenile form of Tay-Sachs disease is less common. Signs and symptoms vary in severity and begin in childhood. Survival is typically into the teen years. Signs and symptoms can include:
- Behavior problems
- Gradual loss of skills and movement control
- Frequent respiratory infections
- Slow loss of vision and speech
- Decline in mental function and responsiveness
Last onset/adult form
This is a rare and less severe form with signs and symptoms beginning in late childhood to adulthood. Severity of symptoms varies greatly, and this form does not always impact life expectancy. Signs and symptoms progress slowly and can include:
- Muscle weakness
- Clumsiness and loss of coordination
- Tremors and muscle spasms
- Loss of the ability to walk
- Problems speaking and swallowing
- Psychiatric disorders
- Sometimes loss of mental function
When to see a doctor
If you or your child has any of the signs or symptoms that may indicate Tay-Sachs disease, or if you have concerns about your child's development, schedule an appointment with your health care provider.
Tay-Sachs disease is a genetic disorder that is passed from parents to their children. It occurs when a child inherits a flaw (mutation) in the HEXA gene from both parents.
The genetic change that causes Tay-Sachs disease results in a deficiency of the enzyme beta-hexosaminidase A. This enzyme is required to break down the fatty substance GM2 ganglioside. The buildup of fatty substances damages nerve cells in the brain and spinal cord. Severity and age of onset of the disease relates to how much enzyme is still produced.
Risk factors
Because the gene change that causes Tay-Sachs disease is found more often in certain populations, risk factors for Tay-Sachs disease include having ancestors from:
- Eastern and Central European Jewish communities (Ashkenazi Jews)
- Certain French Canadian communities in Quebec
- Cajun community of Louisiana
- Old Order Amish community in Pennsylvania
A blood test can be used to identify carriers of the HEXA gene change that causes Tay-Sachs disease. Genetic counseling is recommended following testing.
Tay-Sachs disease care at Mayo Clinic
- Tay-Sachs disease. Genetic and Rare Diseases Information Center. https://rarediseases.info.nih.gov/diseases/7737/tay-sachs-disease. Accessed July 9, 2021.
- About Tay-Sachs disease. National Human Genome Research Institute. https://www.genome.gov/Genetic-Disorders/Tay-Sachs-Disease. Accessed July 9, 2021.
- Vasquez KL. Tay-Sachs disease. Journal of Neonatal Nursing. 2020; doi:10.1016/j.jnn.2020.02.001.
- Solovyeva VV, et al. New approaches to Tay-Sachs disease therapy. Frontiers in Physiology. 2018; doi:10.3389/fphys.2018.01663.
- Tay-Sachs disease information page. National Institute of Neurological Disorders and Stroke. https://www.ninds.nih.gov/Disorders/All-Disorders/Tay-Sachs-Disease-Information-Page. Accessed July 9, 2021.
- Pichurin PN (expert opinion). Mayo Clinic. Nov. 12, 2021.
- Symptoms & causes
- Diagnosis & treatment
- Doctors & departments
- Care at Mayo Clinic
Mayo Clinic does not endorse companies or products. Advertising revenue supports our not-for-profit mission.
- Opportunities
Mayo Clinic Press
Check out these best-sellers and special offers on books and newsletters from Mayo Clinic Press .
- Mayo Clinic on Incontinence - Mayo Clinic Press Mayo Clinic on Incontinence
- The Essential Diabetes Book - Mayo Clinic Press The Essential Diabetes Book
- Mayo Clinic on Hearing and Balance - Mayo Clinic Press Mayo Clinic on Hearing and Balance
- FREE Mayo Clinic Diet Assessment - Mayo Clinic Press FREE Mayo Clinic Diet Assessment
- Mayo Clinic Health Letter - FREE book - Mayo Clinic Press Mayo Clinic Health Letter - FREE book
Your gift holds great power – donate today!
Make your tax-deductible gift and be a part of the cutting-edge research and care that's changing medicine.

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- BMJ Case Rep
Case Report
Tay-sachs disease: a novel mutation from india, daisy khera.
1 Department of Pediatrics, All India Institute of Medical Sciences, Jodhpur, Jodhpur, Rajasthan, India
Joseph John
2 Department of Pediatrics, All India Institute of Medical Sciences Bhubaneswar, Bhubaneswar, Odisha, India
Kuldeep Singh
Mohammed faruq.
3 Genomics and Molecular Medicine, CSIR-Institute of Genomics and Integrative Biology, New Delhi, India
Lysosomal storage disorders or lipidoses are a wide spectrum of inherited diseases caused by deficiency of a specific lysosomal hydrolase. About 134 mutations have been described so far and this number is gradually increasing with newer mutations being reported. We report a 28-month-old child who presented to us with neurodevelopment regression, seizures and cherry red spot in both eyes. His hexosaminidase A enzyme activity was reduced and genetic testing revealed a homozygous novel variation in HEXA (hexosaminidase A) gene in the DNA sample of the patient.
Lysosomal storage disorders (LSDs) are caused by deficiency of a specific lysosomal hydrolase. One of these LSDs is Tay-Sachs disease (TSD) (MIM ID # 272800) which has an autosomal recessive inheritance caused by a genetic mutation in the HEXA gene on human chromosome 15q23–q24. TSD is the second most common lipid storage disorder among the reported lipidoses in India. These reported mutations have included single base pair deletion or multiple base pair insertions, splice site mutations, missense mutations and nonsense mutations. We report a previously unreported missense mutation c.G619C:p.D207H in the HEXA gene in a case of TSD.
Case presentation
A 28-month-old male child, Muslim by religion, presented to us with delayed attainment of developmental milestones and seizures since the latter half of infancy. He was born to second degree consanguineous parents. The mother of the child had history of one spontaneous abortion at 4 months of gestation. This child was born to a 21-year-old, third gravida mother at term gestation, was small for gestational age and weighed 2.2 kg. He required routine care at birth and had normal APGAR scores. His elder sibling was a girl child who was alive and healthy. All seemed well until about 9 months of age after which, a delay in the attainment of newer milestones was noticed by the parents. They were hopeful that he would improve on his own, when at 15 months of age, he had generalised seizures, which made them seek medical attention. After this episode of seizure, he was noticed to have neuroregression in all domains of development. Subsequently, he was on antiepileptic drugs (AEDs) and was on irregular follow-up at various centres but with no significant improvement.
He was brought to our outpatient department at the age of 26 months for evaluation. His vitals were stable and he had severe acute malnutrition on anthropometric examination. His weight was 8.5 kg (less than −3 z score), height was 85 cm (at −2 z score) and weight for height was less than −3 z score. His occipitofrontal circumference was 52 cm which was more than 2 z score for his age suggestive of macrocephaly. He had severe global developmental delay with a developmental age of about 3–4 months. He had no dysmorphism or neurocutaneous stigmata. He was conscious, not focusing or following objects. He had an exaggerated startle response to sound and had no organomegaly. He had symmetrically decreased bulk of muscles, power of 3/5 in all limbs, hypertonia of all limbs with exaggerated deep tendon reflexes and extensor plantar response. His cranial nerve examination was normal. Rest of the systemic examination was unremarkable. His fundus examination revealed bilateral optic atrophy and cherry red spots. His investigations are detailed in table 1 . MRI of the brain showed symmetrical hypomyelination along the internal capsule and bihemispheric white matter ( figure 1 ).
Haematological, biochemical and genetic investigations
ALT, alanine transaminase; ALP, alkaline phosphatase; AST, aspartate transaminase; Hb, haemoglobin; N, neutrophils; PLT, platelet count; TLC, total leucocyte count.

Axial fluid attenuation inversion recovery images showing symmetrical hypomyelination along the internal capsule and bihemispheric white matter.
Differential diagnosis
A provisional diagnosis of TSD/Sandhoff disease was considered.
He was treated with syrup valproic acid and levetiracetam. He was referred to physical medicine and rehabilitation department for multisensory stimulation, physiotherapy and occupational therapy. Prognosis was explained to parents and option of prenatal diagnosis in future pregnancies was explained.
Outcome and follow-up
The child continued to have recurrent seizures and the doses of AEDs were subsequently increased. The child was admitted with lower respiratory tract infection with septic shock to our paediatric intensive care unit at 3.5 years of age and succumbed to the illness. His mother conceived again and was offered prenatal diagnosis. In the fetus, this variation was detected in heterozygous condition. He is now a healthy infant.
Neuroregression in childhood could either be due to genetic causes with neurometabolic origin or non-genetic causes such as infections and toxins. LSDs are one of the causes of neuroregression in children. 1 LSDs are the heritable group of nearly 40 heterogeneous disorders occurring due to genetic defect in one or more specific lysosomal enzymes, activator protein or membrane protein resulting in deficient enzyme activity. 2 3 GM2 gangliosidosis includes TSD and Sandhoff disease. 4 Both types of GM2 gangliosidoses are inherited autosomal recessively with the incidence being as high as 1 in 2500 to 3900 in Ashkenazi Jews. It is relatively rare in India and the exact incidence is not known. Both types are caused from a deficiency of β-hexosaminidase activity and accumulation of GM2 gangliosides mostly in the nervous system or viscera but can also result from defects involved in lysosomal enzyme trafficking or lysosomal enzyme activator proteins. 5 Children with activator protein deficiency, though phenotypically fit into TSD the levels of enzyme activity of hexosaminidase A and total hexosaminidase enzyme activity in the leucocytes would surprisingly be normal. A high index of suspicion is needed in such cases and activator protein deficiency should be looked for. The pathway of metabolism of sphingolipids in nervous tissue and visceral organs is well described. The age of presentation and clinical manifestations of LSDs depend on the amount and rate of intracellular substrates accumulation, the quantum of remaining enzyme which is functional and presence of any alternative functional pathways. 6 Both TSD and Sandhoff disease have infantile, juvenile and adult-onset types based on the onset of symptoms.
β-hexosaminidase has two subunits: β-hexosaminidase A (one α and one β-subunit) and β-hexosaminidase B (two β-subunits). A mutation in the α-subunit gene causes TSD while that in the β-subunit chain gene causes a deficiency of both β-hexosaminidase A and B causing Sandhoff disease. Thus, assessment of β-hexosaminidase enzyme activity in peripheral leucocytes, cultured fibroblasts or lymphoblasts can identify affected individuals and determines the diagnosis. In TSD, only the β-hexosaminidase A isoenzyme is deficient while in Sandhoff disease β-hexosaminidase A and B both isoenzymes are deficient.
TSD is the second most common lipid storage disorder among the majority of LSDs studied in India. 7 The usual manifestations of TSD presenting in infancy are developmental regression, decreased eye contact, exaggerated startle response, macrocephaly not associated with hydrocephalus, macular pallor and a cherry red retinal spot. By the second year of life, the child usually has refractory seizures, relentless neuroregression and death by the fourth or fifth year. The most common causes of morbidity and mortality in LSDs are due to neurological, visceral (cardiovascular and skeletal) accumulation of glycosphingolipids. 8 9 There is no known cure or treatment at present though carrier detection can help in prevention of disease. Prenatal diagnosis is usually done using specific enzyme assay of the chorionic villus sample or cultured amniocytes. But this may give erroneous results due to problems with regard to technical expertise, sample transportation and maternal tissue contamination. If the mutation is identified in the proband or in the carrier parents, a targeted mutation analysis in the fetal DNA can also help diagnose the disease in the fetus. A combination of molecular genetic testing along with enzyme assay has been found to significantly increase the reliability of the prenatal diagnostic procedure 10 and guide genetic counselling. The limitation with molecular genetic testing is the limited availability of advanced centres for such testing and the high cost.
There are over 130 mutations described in TSD, most being in the infantile variety. 11 The most commonly reported ones are the missense mutations (1) c.532CNT (p.R178C), (2) c.964GNT (p.D322Y), (3) c.1385ANT (p.E462V), (4) c.1277_1278insTATC (5) p.D175A (6) p.G269R; the nonsense mutation p.R510X, splice site mutation c.459+5G>A and deletions (1 bp deletion c.425deIT). 9
Learning points
- All children with a history of neurodevelopment regression should undergo fundus examination for cherry red spots and should be evaluated for abnormal startle response to noise and if present should be investigated for hexosaminidase enzyme activity.
- Genetic testing of such cases is important to establish the diagnosis.
- Genetic counselling and prenatal diagnosis should be offered to the parents of such cases.
Acknowledgments
We would like to acknowledge MLP1601, Genomics and other omics tools for enabling Medical Decision (GOMED) project from CSIR for funding the genetic analysis in this case.
Patient consent for publication: Parental/guardian consent obtained.
Contributors: DK made substantial contributions to the conception of the work, drafting the work, revising it critically for important intellectual content and final approval of the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. JJ made substantial contributions to the conception of the work, drafting the work, revising it critically for important intellectual content and final approval of the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. KS made substantial contributions to the conception of the work, drafting the work, revising it critically for important intellectual content and final approval of the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. MF made substantial contributions to the drafting of the work, revising it critically for important intellectual content and final approval of the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
IMAGES
VIDEO
COMMENTS
Tay Sachs disease (TSD) is a progressive, lethal neurodegenerative disorder caused by a deficiency of enzyme hexosaminidase-A resulting in the accumulation of GM2 gangliosides. Based on the presentation age, the disease is classified into infantile, juvenile, and adult forms. Early diagnosis of Tay Sachs is clinically challenging because of subtle clinical features and nonspecific biochemical ...
Tay-Sachs disease is considered a prototype disease for tar geted ethnic. evaluations. (5) TSD occurs most often in children with intellectual disability, skill. regression, dementia, paralysis ...
Abstract. Tay-Sachs disease belongs to the group of autosomal-recessive lysosomal storage metabolic disorders. This disease is caused by β-hexosaminidase A (HexA) enzyme deficiency due to various mutations in α-subunit gene of this enzyme, resulting in GM2 ganglioside accumulation predominantly in lysosomes of nerve cells.
Tay-Sachs disease is a rare inherited neurodegenerative disorder that destroys neurons in the brain and spinal cord in a progressive manner ( Genetics and Rare Diseases Information Center, 2018 ). TSD was named after Warren Tay and Bernard Sachs. Tay, an ophthalmologist, was the first person to describe a cherry-red spot on the retina of a patient.
BioDrugs (2023) Tay-Sachs disease (TSD) is an inherited neurological disorder caused by deficiency of hexosaminidase A (HexA). Here, we describe an adeno-associated virus (AAV) gene therapy ...
Tay-Sachs disease ... The Strategic Priority Research Program of the Chinese Academy of Sciences (XDA16030501, XDA16030503), Key Research & Development Program of Guangzhou Regenerative Medicine ...
Tay-Sachs disease (TSD) is an inherited condition caused by mutations in the genes encoding β-hexosaminidase A (HexA), an enzyme responsible for the recycling of GM2 gangliosides ...
Tay-Sachs disease (TSD) is a rare neurodegenerative disorder caused by mutations in the HEXA gene, which encodes the ɑ subunit of the enzyme β-hexosaminidase A, which results in the progressive deterioration of the central nervous system. Tay-Sachs disease (TSD) is a rare neurodegenerative disorder caused by mutations in the HEXA gene, which encodes the ɑ subunit of the enzyme β ...
Named after ophthalmologist Warren Tay and neurologist Bernard Sachs, Tay-Sachs disease (TSD) is a neurodegenerative disorder caused by the excessive accumulation of gangliosides in the nerve cells of the central nervous system 1.Following Tay's 1881 description of "a conspicuous, tolerably defined, large white patch, more or less circular in outline, and showing at its centre a brownish ...
Please use one of the following formats to cite this article in your essay, paper or report: APA. Smith, Yolanda. (2021, May 17). Tay-Sachs Disease Research.
Tay-Sachs disease (TSD) is a fatal, recessively inherited neurodegenerative condition of infancy and early childhood. Although rare in most other populations, the carrier frequency is one in 25 in Ashkenazi Jews. Australian high-school-based TSD preconception genetic screening programs aim to screen, educate, and optimize reproductive choice ...
Tay-Sachs disease (TSD) is a recessively inherited neurodegenerative disorder caused by mutations in the HEXA gene resulting in β-hexosaminidase A (HEX A) deficiency and neuronal accumulation of GM2 ganglioside. We describe the first patient with Tay-Sachs disease in the Cypriot population, a juvenile case which presented with developmental regression at the age of five.
Tay-Sachs disease is an autosomal recessive disease caused by a deficiency of β-hexosaminidase A, the lysosomal enzyme that normally degrades GM2 ganglioside. ... in his landmark paper Sachs acknowledged Adler as having sent him his first case of what would later be known as infantile Tay-Sachs disease. ... "All true scientific research in ...
Tay-Sachs disease (TSD) is a fatal genetic disorder, most commonly occurring in children, that results in progressive destruction of the nervous system. Tay-Sachs is caused by the absence of a vital enzyme called hexosaminidase-A (Hex-A). Without Hex-A, a fatty substance, or lipid, called GM2 ganglioside accumulates abnormally in cells ...
The National Institutes of Health (NIH) recently awarded the Tay-Sachs Gene Therapy Consortium (Consortium) a four-year, $3.5 million grant to continue its cutting-edge research to cure this fatal disease. The Consortium, formed in March 2007, is an international collaborative group of scientists from four institutions: Auburn University ...
Tay-Sachs disease is a genetic disease that has been at the forefront of scientific research on inheritance patterns. ... paper or report: APA. Smith, Yolanda. (2022, September 26).
Tay-Sachs disease (TSD) is a fatal neurodegenerative disease caused by a deficiency of the enzyme β-N-acetylhexosaminidase A (HexA). TSD naturally occurs in Jacob sheep is the only experimental model of TSD. TSD in sheep recapitulates neurologic features similar to juvenile onset and late onset TSD patients. Due to the paucity of human ...
Disease Researchers. Specialists who have done research into Tay-Sachs disease. These specialists have recieved grants, written articles, run clinical trials, or taken part in organizations relating to Tay-Sachs disease, and are considered knowledgeable about the disease as a result. The people in this list are filtered based on their research ...
The juvenile form of Tay-Sachs disease is less common. Signs and symptoms vary in severity and begin in childhood. Survival is typically into the teen years. Signs and symptoms can include: Behavior problems. Gradual loss of skills and movement control. Frequent respiratory infections. Slow loss of vision and speech.
Background. Lysosomal storage disorders (LSDs) are caused by deficiency of a specific lysosomal hydrolase. One of these LSDs is Tay-Sachs disease (TSD) (MIM ID # 272800) which has an autosomal recessive inheritance caused by a genetic mutation in the HEXA gene on human chromosome 15q23-q24. TSD is the second most common lipid storage disorder ...